Related Research Articles

A lysosome is a single membrane-bound organelle found in many animal cells. They are spherical vesicles that contain hydrolytic enzymes that digest many kinds of biomolecules. A lysosome has a specific composition, of both its membrane proteins and its lumenal proteins. The lumen's pH (~4.5–5.0) is optimal for the enzymes involved in hydrolysis, analogous to the activity of the stomach. Besides degradation of polymers, the lysosome is involved in cell processes of secretion, plasma membrane repair, apoptosis, cell signaling, and energy metabolism.

Lysosomal storage diseases are a group of over 70 rare inherited metabolic disorders that result from defects in lysosomal function. Lysosomes are sacs of enzymes within cells that digest large molecules and pass the fragments on to other parts of the cell for recycling. This process requires several critical enzymes. If one of these enzymes is defective due to a mutation, the large molecules accumulate within the cell, eventually killing it.

Niemann–Pick disease (NP), also known as acid sphingomyelinase deficiency, is a group of rare genetic diseases of varying severity. These are inherited metabolic disorders in which sphingomyelin accumulates in lysosomes in cells of many organs. NP types A, A/B, and B are caused by mutations in the SMPD1 gene, which causes a deficiency of an acid sphingomyelinase (ASM). NP type C is now considered a separate disease, as SMPD1 is not involved, and there is no deficiency in ASM.
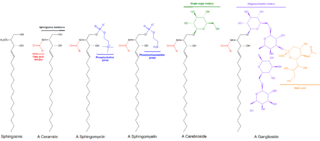
Sphingolipids are a class of lipids containing a backbone of sphingoid bases, which are a set of aliphatic amino alcohols that includes sphingosine. They were discovered in brain extracts in the 1870s and were named after the mythological sphinx because of their enigmatic nature. These compounds play important roles in signal transduction and cell recognition. Sphingolipidoses, or disorders of sphingolipid metabolism, have particular impact on neural tissue. A sphingolipid with a terminal hydroxyl group is a ceramide. Other common groups bonded to the terminal oxygen atom include phosphocholine, yielding a sphingomyelin, and various sugar monomers or dimers, yielding cerebrosides and globosides, respectively. Cerebrosides and globosides are collectively known as glycosphingolipids.
Sphingomyelin is a type of sphingolipid found in animal cell membranes, especially in the membranous myelin sheath that surrounds some nerve cell axons. It usually consists of phosphocholine and ceramide, or a phosphoethanolamine head group; therefore, sphingomyelins can also be classified as sphingophospholipids. In humans, SPH represents ~85% of all sphingolipids, and typically make up 10–20 mol % of plasma membrane lipids.

The low-density lipoprotein receptor (LDL-R) is a mosaic protein of 839 amino acids that mediates the endocytosis of cholesterol-rich low-density lipoprotein (LDL). It is a cell-surface receptor that recognizes apolipoprotein B100 (ApoB100), which is embedded in the outer phospholipid layer of very low-density lipoprotein (VLDL), their remnants—i.e. intermediate-density lipoprotein (IDL), and LDL particles. The receptor also recognizes apolipoprotein E (ApoE) which is found in chylomicron remnants and IDL. In humans, the LDL receptor protein is encoded by the LDLR gene on chromosome 19. It belongs to the low density lipoprotein receptor gene family. It is most significantly expressed in bronchial epithelial cells and adrenal gland and cortex tissue.

Ceramides are a family of waxy lipid molecules. A ceramide is composed of sphingosine and a fatty acid joined by an amide bond. Ceramides are found in high concentrations within the cell membrane of eukaryotic cells, since they are component lipids that make up sphingomyelin, one of the major lipids in the lipid bilayer. Contrary to previous assumptions that ceramides and other sphingolipids found in cell membrane were purely supporting structural elements, ceramide can participate in a variety of cellular signaling: examples include regulating differentiation, proliferation, and programmed cell death (PCD) of cells.
Farber disease is an extremely rare, progressive, autosomal recessive lysosomal storage disease caused by a deficiency of the acid ceramidase enzyme. Acid ceramidase is responsible for breaking down ceramide into sphingosine and fatty acid. When the enzyme is deficient, this leads to an accumulation of fatty material in the lysosomes of the cells, leading to the signs and symptoms of this disorder.

β-Glucocerebrosidase is an enzyme with glucosylceramidase activity that cleaves by hydrolysis the β-glycosidic linkage of the chemical glucocerebroside, an intermediate in glycolipid metabolism that is abundant in cell membranes. It is localized in the lysosome, where it remains associated with the lysosomal membrane. β-Glucocerebrosidase is 497 amino acids in length and has a molecular mass of 59,700 Da.

Lipid signaling, broadly defined, refers to any biological cell signaling event involving a lipid messenger that binds a protein target, such as a receptor, kinase or phosphatase, which in turn mediate the effects of these lipids on specific cellular responses. Lipid signaling is thought to be qualitatively different from other classical signaling paradigms because lipids can freely diffuse through membranes. One consequence of this is that lipid messengers cannot be stored in vesicles prior to release and so are often biosynthesized "on demand" at their intended site of action. As such, many lipid signaling molecules cannot circulate freely in solution but, rather, exist bound to special carrier proteins in serum.

Sphingomyelin phosphodiesterase is a hydrolase enzyme that is involved in sphingolipid metabolism reactions. SMase is a member of the DNase I superfamily of enzymes and is responsible for breaking sphingomyelin (SM) down into phosphocholine and ceramide. The activation of SMase has been suggested as a major route for the production of ceramide in response to cellular stresses.
Acid lipase disease or deficiency is a name used to describe two related disorders of fatty acid metabolism. Acid lipase disease occurs when the enzyme lysosomal acid lipase that is needed to break down certain fats that are normally digested by the body is lacking or missing. This results in the toxic buildup of these fats in the body's cells and tissues. These fatty substances, called lipids, include waxes, oils, and cholesterol.
In enzymology, a ceramide kinase, also abbreviated as CERK, is an enzyme that catalyzes the chemical reaction:

Sphingomyelin phosphodiesterase 1 (SMPD1), also known as acid sphingomyelinase (ASM), is an enzyme that in humans is encoded by the SMPD1 gene.

The ASAH1 gene encodes in humans the acid ceramidase enzyme.

Ectonucleotide pyrophosphatase/phosphodiesterase family member 7 also known as alkaline sphingomyelin phosphodiesterase (Alk-SMase) or intestinal alkaline sphingomyelinase is an enzyme that in humans is encoded by the ENPP7 gene.

Niemann–Pick type C (NPC) is a lysosomal storage disease associated with mutations in NPC1 and NPC2 genes. Niemann–Pick type C affects an estimated 1:150,000 people. Approximately 50% of cases present before 10 years of age, but manifestations may first be recognized as late as the sixth decade.
SMPD1-associated Niemann–Pick disease refers to two different types of Niemann–Pick disease, type A (NPA) and type B (NPB), which are associated with the SMPD1 gene.

Robert J. Desnick is an American human geneticist whose basic and translational research accomplishments include significant discoveries in genomics, pharmacogenetics, gene therapy, personalized medicine, and the treatment of genetic diseases. His translational research has led to the development of the enzyme replacement therapy (ERT) and the chaperone therapy for Fabry disease, ERT for Niemann–Pick disease type B, and the RNA Interference Therapy for the Acute Hepatic Porphyrias.
Functional inhibitors of acid sphingomyelinase, or FIASMA, is a large group of pharmacological compounds inhibiting the enzyme acid sphingomyelinase. This enzyme is mainly located within the lysosome, where it cleaves sphingomyelin to ceramide and sphingosine, the latter of which is then phosphorylated to sphingosine-1-phosphate. These metabolites, and subsequent inhibition of the enzyme, influence the balance between cell death (apoptosis) and cell growth (proliferation). A lack of regulation of this sensitive equilibrium can lead to serious clinical consequences.
References
- 1 2 Schuchman, Edward H (May 2010). "Acid Sphingomyelinase, cell membranes and human disease: Lessons from Niemann-Pick disease". FEBS Letters. 584 (9): 1895–1900. Bibcode:2010FEBSL.584.1895S. doi: 10.1016/j.febslet.2009.11.083 . PMID 19944693.
- ↑ Kornhuber J, Muehlbacher M, Trapp S, Pechmann S, Friedl A, Reichel M, Muehle C, Terfloth L, Groemer TW, Spitzer GM, Liedl KR, Gulbins E, Tribal P (2006). "Identification of novel functional inhibitors of acid sphingomyelinase". PLOS ONE. 6 (8): e23852. doi: 10.1371/journal.pone.0023852 . PMC 3166082 . PMID 21909365.
- 1 2 McGovern, Mm; Schuchman EH (7 Dec 2006). "Acid Sphingomyelinase Deficiency". GeneReviews.
- 1 2 3 4 5 6 7 Jenkins, RW; Canals D; Hannun YA (21 June 2009). "Roles and Regulation of Secretory and Lysosomal Acid Sphingomyelinases". Cell Signal. 21 (6): 836–846. doi:10.1016/j.cellsig.2009.01.026. PMC 3488588 . PMID 19385042.
- ↑ "BIS(MONOACYLGLYCERO)PHOSPHATE structure, occurrence, and biochemistry". the AOCS Lipid Library. Archived from the original on 2013-06-06.
- ↑ Cowart, Ashley (2011). Sphingolipids and Metabolic Disease. Springer. pp. 71–152. ISBN 978-1-4614-0650-1.
- ↑ Kornhuber, Johannes; Hoertel, Nicolas; Gulbins, Erich (Oct 2021). "The acid sphingomyelinase/ceramide system in COVID-19". Molecular Psychiatry. 27 (1): 307–314. doi:10.1038/s41380-021-01309-5. ISSN 1476-5578. PMC 8488928 . PMID 34608263.
- ↑ Sánchez-Rico, Marina; Limosin, Frédéric; Vernet, Raphaël; Beeker, Nathanaël; Neuraz, Antoine; Blanco, Carlos; Olfson, Mark; Lemogne, Cédric; Meneton, Pierre; Daniel, Christel; Paris, Nicolas; Gramfort, Alexandre; Lemaitre, Guillaume; De La Muela, Pedro; Salamanca, Elisa (2021-12-15). "Hydroxyzine Use and Mortality in Patients Hospitalized for COVID-19: A Multicenter Observational Study". Journal of Clinical Medicine. 10 (24): 5891. doi: 10.3390/jcm10245891 . ISSN 2077-0383. PMC 8707307 . PMID 34945186.