 | |
| Clinical data | |
|---|---|
| Other names | LY-293,558; LY-293558; LY293558; LY-326,325; LY-326325; LY326325; NGX-424; NGX424 |
| Routes of administration | IV |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
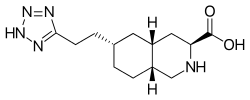
| Formula | C13H21N5O2 |
| Molar mass | 279.344 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
| | |
Tezampanel (INN , USAN ) (developmental code names LY-293,558, LY-326,325, NGX-424) is a drug originally developed by Eli Lilly [1] which acts as a competitive antagonist of the AMPA and kainate subtypes of the ionotropic glutamate receptor family, [2] [3] with selectivity for the GluR5 subtype of the kainate receptor. [4] [5] It has neuroprotective [6] and anticonvulsant properties, [7] the former of which may, at least in part, occur via blockade of calcium uptake into neurons. [8]
Tezampanel has a range of effects which may be useful for medicinal purposes, as well as its applications in scientific research. It suppresses both the withdrawal symptoms from morphine and other opioids, [9] [10] [11] and the development of tolerance, [12] as well as having antihyperalgesic [13] and analgesic effects in its own right. [14] [15] [16] [17] [18] It also has anxiolytic effects in animal studies and has been suggested as a candidate for the treatment of anxiety in humans. [19]
Whereas tezampanel free base is known as LY-293558, tezampanel hydrochloride is said to be known as LY-326325. [20] [21]