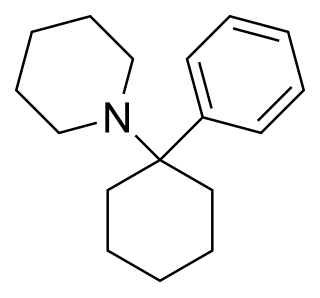
Phencyclidine or phenylcyclohexyl piperidine (PCP), also known in its use as a street drug as angel dust among other names, is a dissociative anesthetic mainly used recreationally for its significant mind-altering effects. PCP may cause hallucinations, distorted perceptions of sounds, and violent behavior. As a recreational drug, it is typically smoked, but may be taken by mouth, snorted, or injected. It may also be mixed with cannabis or tobacco.
A dopamine reuptake inhibitor (DRI) is a class of drug which acts as a reuptake inhibitor of the monoamine neurotransmitter dopamine by blocking the action of the dopamine transporter (DAT). Reuptake inhibition is achieved when extracellular dopamine not absorbed by the postsynaptic neuron is blocked from re-entering the presynaptic neuron. This results in increased extracellular concentrations of dopamine and increase in dopaminergic neurotransmission.

Dizocilpine (INN), also known as MK-801, is a pore blocker of the NMDA receptor, a glutamate receptor, discovered by a team at Merck in 1982. Glutamate is the brain's primary excitatory neurotransmitter. The channel is normally blocked with a magnesium ion and requires depolarization of the neuron to remove the magnesium and allow the glutamate to open the channel, causing an influx of calcium, which then leads to subsequent depolarization. Dizocilpine binds inside the ion channel of the receptor at several of PCP's binding sites thus preventing the flow of ions, including calcium (Ca2+), through the channel. Dizocilpine blocks NMDA receptors in a use- and voltage-dependent manner, since the channel must open for the drug to bind inside it. The drug acts as a potent anti-convulsant and probably has dissociative anesthetic properties, but it is not used clinically for this purpose because of the discovery of brain lesions, called Olney's lesions (see below), in laboratory rats. Dizocilpine is also associated with a number of negative side effects, including cognitive disruption and psychotic-spectrum reactions. It inhibits the induction of long term potentiation and has been found to impair the acquisition of difficult, but not easy, learning tasks in rats and primates. Because of these effects of dizocilpine, the NMDA receptor pore blocker ketamine is used instead as a dissociative anesthetic in human medical procedures. While ketamine may also trigger temporary psychosis in certain individuals, its short half-life and lower potency make it a much safer clinical option. However, dizocilpine is the most frequently used uncompetitive NMDA receptor antagonist in animal models to mimic psychosis for experimental purposes.
Neuropharmacology is the study of how drugs affect function in the nervous system, and the neural mechanisms through which they influence behavior. There are two main branches of neuropharmacology: behavioral and molecular. Behavioral neuropharmacology focuses on the study of how drugs affect human behavior (neuropsychopharmacology), including the study of how drug dependence and addiction affect the human brain. Molecular neuropharmacology involves the study of neurons and their neurochemical interactions, with the overall goal of developing drugs that have beneficial effects on neurological function. Both of these fields are closely connected, since both are concerned with the interactions of neurotransmitters, neuropeptides, neurohormones, neuromodulators, enzymes, second messengers, co-transporters, ion channels, and receptor proteins in the central and peripheral nervous systems. Studying these interactions, researchers are developing drugs to treat many different neurological disorders, including pain, neurodegenerative diseases such as Parkinson's disease and Alzheimer's disease, psychological disorders, addiction, and many others.

NMDA receptor antagonists are a class of drugs that work to antagonize, or inhibit the action of, the N-Methyl-D-aspartate receptor (NMDAR). They are commonly used as anesthetics for humans and animals; the state of anesthesia they induce is referred to as dissociative anesthesia.
(+)-CPCA is a stimulant drug similar in structure to pethidine and to RTI-31, but nocaine lacks the two-carbon bridge of RTI-31's tropane skeleton. This compound was first developed as a substitute agent for cocaine.

Indalpine, sold under the brand name Upstène, is a selective serotonin reuptake inhibitor (SSRI) that was briefly marketed as an antidepressant for treatment of depression. It was marketed in France and a few other European countries.

Femoxetine is a drug related to paroxetine that was being developed as an antidepressant by Danish pharmaceutical company Ferrosan in 1975 before acquisition of the company by Novo Nordisk. It acts as a selective serotonin reuptake inhibitor (SSRI). Development was halted to focus attention on paroxetine instead, as femoxetine could not be administered as a daily pill.
Etoxadrol (CL-1848C) is a dissociative anaesthetic drug that has been found to be an NMDA antagonist and produce similar effects to PCP in animals. Etoxadrol, along with another related drug dexoxadrol, were developed as analgesics for use in humans, but development was discontinued in the late 1970s after patients reported side effects such as nightmares and hallucinations.
Selfotel (CGS-19755) is a drug which acts as a competitive NMDA antagonist, directly competing with glutamate for binding to the receptor. Initial studies showed it to have anticonvulsant, anxiolytic, analgesic and neuroprotective effects, and it was originally researched for the treatment of stroke, but subsequent animal and human studies showed phencyclidine-like effects, as well as limited efficacy and evidence for possible neurotoxicity under some conditions, and so clinical development was ultimately discontinued.

A serotonin releasing agent (SRA) is a type of drug that induces the release of serotonin into the neuronal synaptic cleft. A selective serotonin releasing agent (SSRA) is an SRA with less significant or no efficacy in producing neurotransmitter efflux at other types of monoamine neurons, including dopamine and norepinephrine neurons.

Litoxetine (developmental code names SL 81-0385, IXA-001) is an antidepressant which was under clinical development for the treatment of depression in the early 1990s but was never marketed. It acts as a potent serotonin reuptake inhibitor (Ki for SERTTooltip serotonin transporter = 7 nM) and modest 5-HT3 receptor antagonist (Ki = 315 nM). It has antiemetic activity, and unlike the selective serotonin reuptake inhibitors (SSRIs), appears to have a negligible incidence of nausea and vomiting. The drug is structurally related to indalpine. Development of litoxetine for depression was apparently ceased in the late 1990s. However, as of March 2017, development of litoxetine has been reinitiated and the drug is now in the phase II stage for the treatment of urinary incontinence.
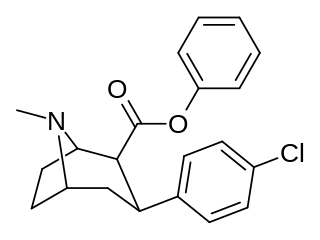
RTI(-4229)-113 is a stimulant drug which acts as a potent and fully selective dopamine reuptake inhibitor (DRI). It has been suggested as a possible substitute drug for the treatment of cocaine addiction. "RTI-113 has properties that make it an ideal medication for cocaine abusers, such as an equivalent efficacy, a higher potency, and a longer duration of action as compared to cocaine." Replacing the methyl ester in RTI-31 with a phenyl ester makes the resultant RTI-113 fully DAT specific. RTI-113 is a particularly relevant phenyltropane cocaine analog that has been tested on squirrel monkeys. RTI-113 has also been tested against cocaine in self-administration studies for DAT occupancy by PET on awake rhesus monkeys. The efficacy of cocaine analogs to elicit self-administration is closely related to the rate at which they are administered. Slower onset of action analogs are less likely to function as positive reinforcers than analogues that have a faster rate of onset.
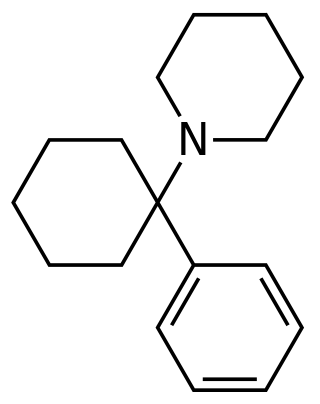
Arylcyclohexylamines, also known as arylcyclohexamines or arylcyclohexanamines, are a chemical class of pharmaceutical, designer, and experimental drugs.

Dextrallorphan (DXA) is a chemical of the morphinan class that is used in scientific research. It acts as a σ1 receptor agonist and NMDA receptor antagonist. It has no significant affinity for the σ2, μ-opioid, or δ-opioid receptor, or for the serotonin or norepinephrine transporter. As an NMDA receptor antagonist, in vivo, it is approximately twice as potent as dextromethorphan, and five-fold less potent than dextrorphan.

Traxoprodil is a drug developed by Pfizer which acts as an NMDA antagonist, selective for the NR2B subunit. It has neuroprotective, analgesic, and anti-Parkinsonian effects in animal studies. Traxoprodil has been researched in humans as a potential treatment to lessen the damage to the brain after stroke, but results from clinical trials showed only modest benefit. The drug was found to cause EKG abnormalities and its clinical development was stopped. More recent animal studies have suggested traxoprodil may exhibit rapid-acting antidepressant effects similar to those of ketamine, although there is some evidence for similar psychoactive side effects and abuse potential at higher doses, which might limit clinical acceptance of traxoprodil for this application.
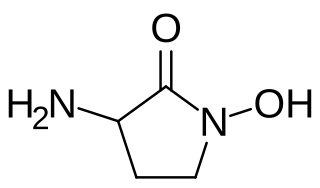
HA-966 or (±)-3-amino-1-hydroxy-pyrrolidin-2-one is a molecule used in scientific research as a glycine receptor and NMDA receptor antagonist / low efficacy partial agonist. It has neuroprotective and anticonvulsant, anxiolytic, antinociceptive and sedative / hypnotic effects in animal models. Pilot human clinical trials in the early 1960s showed that HA-966 appeared to benefit patients with tremors of extrapyramidal origin.
Selective serotonin reuptake inhibitors, or serotonin-specific re-uptake inhibitor (SSRIs), are a class of chemical compounds that have application as antidepressants and in the treatment of depression and other psychiatric disorders. SSRIs are therapeutically useful in the treatment of panic disorder (PD), posttraumatic stress disorder (PTSD), social anxiety disorder, obsessive-compulsive disorder (OCD), premenstrual dysphoric disorder (PMDD), and anorexia. There is also clinical evidence of the value of SSRIs in the treatment of the symptoms of schizophrenia and their ability to prevent cardiovascular diseases.
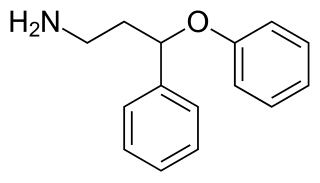
PPPA, or 3-phenoxy-3-phenylpropan-1-amine, is a drug which is described as an antidepressant. It was derived by Eli Lilly from the antihistamine diphenhydramine, a diphenylmethane derivative with additional properties as a selective serotonin reuptake inhibitor (SSRI), and has been the basis for the subsequent discovery of a number of other antidepressant drugs.

PD-137889 (N-methylhexahydrofluorenamine) is a chemical compound that is active as an NMDA receptor antagonist in the central nervous system at roughly 30 times the potency of the "flagship" of its class, ketamine, and substitutes for phencyclidine in animal studies. Ki [3H]TCP binding = 27 nM versus ketamine's Ki = 860 nM.

