
The α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (also known as AMPA receptor, AMPAR, or quisqualate receptor) is an ionotropic transmembrane receptor for glutamate (iGluR) and predominantly Na+ ion channel that mediates fast synaptic transmission in the central nervous system (CNS). It has been traditionally classified as a non-NMDA-type receptor, along with the kainate receptor. Its name is derived from its ability to be activated by the artificial glutamate analog AMPA. The receptor was first named the "quisqualate receptor" by Watkins and colleagues after a naturally occurring agonist quisqualate and was only later given the label "AMPA receptor" after the selective agonist developed by Tage Honore and colleagues at the Royal Danish School of Pharmacy in Copenhagen. The GRIA2-encoded AMPA receptor ligand binding core (GluA2 LBD) was the first glutamate receptor ion channel domain to be crystallized.

The N-methyl-D-aspartatereceptor (also known as the NMDA receptor or NMDAR), is a glutamate receptor and predominantly Ca2+ ion channel found in neurons. The NMDA receptor is one of three types of ionotropic glutamate receptors, the other two being AMPA and kainate receptors. Depending on its subunit composition, its ligands are glutamate and glycine (or D-serine). However, the binding of the ligands is typically not sufficient to open the channel as it may be blocked by Mg2+ ions which are only removed when the neuron is sufficiently depolarized. Thus, the channel acts as a "coincidence detector" and only once both of these conditions are met, the channel opens and it allows positively charged ions (cations) to flow through the cell membrane. The NMDA receptor is thought to be very important for controlling synaptic plasticity and mediating learning and memory functions.
In neurophysiology, long-term depression (LTD) is an activity-dependent reduction in the efficacy of neuronal synapses lasting hours or longer following a long patterned stimulus. LTD occurs in many areas of the CNS with varying mechanisms depending upon brain region and developmental progress.

Ibotenic acid or (S)-2-amino-2-(3-hydroxyisoxazol-5-yl)acetic acid, also referred to as ibotenate, is a chemical compound and psychoactive drug which occurs naturally in Amanita muscaria and related species of mushrooms typically found in the temperate and boreal regions of the northern hemisphere. It is a prodrug of muscimol, broken down by the liver to that much more stable compound. It is a conformationally-restricted analogue of the neurotransmitter glutamate, and due to its structural similarity to this neurotransmitter, acts as a non-selective glutamate receptor agonist. Because of this, ibotenic acid can be a powerful neurotoxin in high doses, and is employed as a "brain-lesioning agent" through cranial injections in scientific research. The neurotoxic effects appear to be dose-related and risks are unclear through consumption of ibotenic-acid containing fungi, although thought to be negligible in small doses.

Ligand-gated ion channels (LICs, LGIC), also commonly referred to as ionotropic receptors, are a group of transmembrane ion-channel proteins which open to allow ions such as Na+, K+, Ca2+, and/or Cl− to pass through the membrane in response to the binding of a chemical messenger (i.e. a ligand), such as a neurotransmitter.

Kainate receptors, or kainic acid receptors (KARs), are ionotropic receptors that respond to the neurotransmitter glutamate. They were first identified as a distinct receptor type through their selective activation by the agonist kainate, a drug first isolated from the algae Digenea simplex. They have been traditionally classified as a non-NMDA-type receptor, along with the AMPA receptor. KARs are less understood than AMPA and NMDA receptors, the other ionotropic glutamate receptors. Postsynaptic kainate receptors are involved in excitatory neurotransmission. Presynaptic kainate receptors have been implicated in inhibitory neurotransmission by modulating release of the inhibitory neurotransmitter GABA through a presynaptic mechanism.
Kainic acid, or kainate, is an acid that naturally occurs in some seaweed. Kainic acid is a potent neuroexcitatory amino acid agonist that acts by activating receptors for glutamate, the principal excitatory neurotransmitter in the central nervous system. Glutamate is produced by the cell's metabolic processes and there are four major classifications of glutamate receptors: NMDA receptors, AMPA receptors, kainate receptors, and the metabotropic glutamate receptors. Kainic acid is an agonist for kainate receptors, a type of ionotropic glutamate receptor. Kainate receptors likely control a sodium channel that produces excitatory postsynaptic potentials (EPSPs) when glutamate binds.

The metabotropic glutamate receptors, or mGluRs, are a type of glutamate receptor that are active through an indirect metabotropic process. They are members of the group C family of G-protein-coupled receptors, or GPCRs. Like all glutamate receptors, mGluRs bind with glutamate, an amino acid that functions as an excitatory neurotransmitter.

Glutamate receptors are synaptic and non synaptic receptors located primarily on the membranes of neuronal and glial cells. Glutamate is abundant in the human body, but particularly in the nervous system and especially prominent in the human brain where it is the body's most prominent neurotransmitter, the brain's main excitatory neurotransmitter, and also the precursor for GABA, the brain's main inhibitory neurotransmitter. Glutamate receptors are responsible for the glutamate-mediated postsynaptic excitation of neural cells, and are important for neural communication, memory formation, learning, and regulation.
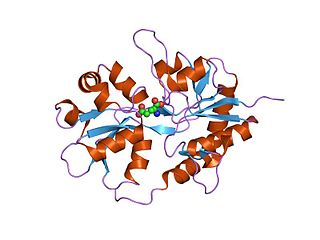
Ionotropic glutamate receptors (iGluRs) are ligand-gated ion channels that are activated by the neurotransmitter glutamate. They mediate the majority of excitatory synaptic transmission throughout the central nervous system and are key players in synaptic plasticity, which is important for learning and memory. iGluRs have been divided into four subtypes on the basis of their ligand binding properties (pharmacology) and sequence similarity: AMPA receptors, kainate receptors, NMDA receptors and delta receptors.

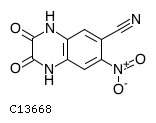
DNQX (6,7-dinitroquinoxaline-2,3-dione) is a competitive antagonist at AMPA and kainate receptors, two ionotropic glutamate receptor (iGluR) subfamilies. It is used in a variety of molecular biology subfields, notably neurophysiology, to assist researchers in determining the properties of various types of ion channels and their potential applications in medicine.

Quisqualic acid is an agonist of the AMPA, kainate, and group I metabotropic glutamate receptors. It is one of the most potent AMPA receptor agonists known. It causes excitotoxicity and is used in neuroscience to selectively destroy neurons in the brain or spinal cord. Quisqualic acid occurs naturally in the seeds of Quisqualis species.

Glutamate ionotropic receptor AMPA type subunit 2 is a protein that in humans is encoded by the GRIA2 gene and it is a subunit found in the AMPA receptors.
The glutamate hypothesis of schizophrenia models the subset of pathologic mechanisms of schizophrenia linked to glutamatergic signaling. The hypothesis was initially based on a set of clinical, neuropathological, and, later, genetic findings pointing at a hypofunction of glutamatergic signaling via NMDA receptors. While thought to be more proximal to the root causes of schizophrenia, it does not negate the dopamine hypothesis, and the two may be ultimately brought together by circuit-based models. The development of the hypothesis allowed for the integration of the GABAergic and oscillatory abnormalities into the converging disease model and made it possible to discover the causes of some disruptions.

Tezampanel is a drug originally developed by Eli Lilly which acts as a competitive antagonist of the AMPA and kainate subtypes of the ionotropic glutamate receptor family, with selectivity for the GluR5 subtype of the kainate receptor. It has neuroprotective and anticonvulsant properties, the former of which may, at least in part, occur via blockade of calcium uptake into neurons.

GYKI 52466 is a 2,3-benzodiazepine that acts as an ionotropic glutamate receptor antagonist, which is a non-competitive AMPA receptor antagonist (IC50 values are 10-20, ~ 450 and >> 50 μM for AMPA-, kainate- and NMDA-induced responses respectively), orally-active anticonvulsant, and skeletal muscle relaxant. Unlike conventional 1,4-benzodiazepines, GYKI 52466 and related 2,3-benzodiazepines do not act on GABAA receptors. Like other AMPA receptor antagonists, GYKI 52466 has anticonvulsant and neuroprotective properties.
In neuroscience, synaptic scaling is a form of homeostatic plasticity, in which the brain responds to chronically elevated activity in a neural circuit with negative feedback, allowing individual neurons to reduce their overall action potential firing rate. Where Hebbian plasticity mechanisms modify neural synaptic connections selectively, synaptic scaling normalizes all neural synaptic connections by decreasing the strength of each synapse by the same factor, so that the relative synaptic weighting of each synapse is preserved.

Quisqualamine is the α-decarboxylated analogue of quisqualic acid, as well as a relative of the neurotransmitters glutamate and γ-aminobutyric acid (GABA). α-Decarboxylation of excitatory amino acids can produce derivatives with inhibitory effects. Indeed, unlike quisqualic acid, quisqualamine has central depressant and neuroprotective properties and appears to act predominantly as an agonist of the GABAA receptor and also to a lesser extent as an agonist of the glycine receptor, due to the facts that its actions are inhibited in vitro by GABAA antagonists like bicuculline and picrotoxin and by the glycine antagonist strychnine, respectively. Mg2+ and DL-AP5, NMDA receptor blockers, CNQX, an antagonist of both the AMPA and kainate receptors, and 2-hydroxysaclofen, a GABAB receptor antagonist, do not affect quisqualamine's actions in vitro, suggesting that it does not directly affect the ionotropic glutamate receptors or the GABAB receptor in any way. Whether it binds to and acts upon any of the metabotropic glutamate receptors like its analogue quisqualic acid however is unclear.

Kaitocephalin is a non-selective ionotropic glutamate receptor antagonist, meaning it blocks the action of the neurotransmitter glutamate. It is produced by the fungus Eupenicillium shearii. Although similar molecules have been produced synthetically, kaitocephalin is the only known naturally occurring glutamate receptor antagonist. There is some evidence that kaitocephalin can protect the brain and central nervous system, so it is said to have neuroprotective properties. Kaitocephalin protects neurons by inhibiting excitotoxicity, a mechanism which causes cell death by overloading neurons with glutamate. Because of this, it is of interest as a potential scaffold for drug development. Drugs based on kaitocephalin may be useful in treating neurological conditions, including Alzheimer's, amyotrophic lateral sclerosis (ALS), and stroke.

Willardiine (correctly spelled with two successive i's) or (S)-1-(2-amino-2-carboxyethyl)pyrimidine-2,4-dione is a chemical compound that occurs naturally in the seeds of Mariosousa willardiana and Acacia sensu lato. The seedlings of these plants contain enzymes capable of complex chemical substitutions that result in the formation of free amino acids (See:#Synthesis). Willardiine is frequently studied for its function in higher level plants. Additionally, many derivates of willardiine are researched for their potential in pharmaceutical development. Willardiine was first discovered in 1959 by R. Gmelin, when he isolated several free, non-protein amino acids from Acacia willardiana (another name for Mariosousa willardiana) when he was studying how these families of plants synthesize uracilyalanines. A related compound, Isowillardiine, was concurrently isolated by a different group, and it was discovered that the two compounds had different structural and functional properties. Subsequent research on willardiine has focused on the functional significance of different substitutions at the nitrogen group and the development of analogs of willardiine with different pharmacokinetic properties. In general, Willardiine is the one of the first compounds studied in which slight changes to molecular structure result in compounds with significantly different pharmacokinetic properties.

{kind=link}