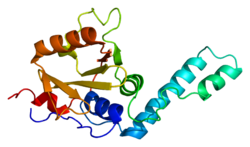




Ataxin-3 is a protein that in humans is encoded by the ATXN3 gene. [5] [6]
Ataxin-3 is a protein that in humans is encoded by the ATXN3 gene. [5] [6]
Machado–Joseph disease, also known as spinocerebellar ataxia-3, is an autosomal dominant neurologic disorder. The protein encoded by the ATXN3 gene contains CAG repeats in the coding region, and the expansion of these repeats from the normal 13-36 to 68-79 is the cause of Machado–Joseph disease. This disorder is thus a trinucleotide repeat disorder type I known as a polyglutamine (PolyQ) disease. There is an inverse correlation between the age of onset and CAG repeat numbers. Alternatively spliced transcript variants encoding different isoforms have been described for this gene. [6]
Ataxin 3 has been shown to interact with:

Spinocerebellar ataxia (SCA) is a progressive, degenerative, genetic disease with multiple types, each of which could be considered a neurological condition in its own right. An estimated 150,000 people in the United States have a diagnosis of spinocerebellar ataxia at any given time. SCA is hereditary, progressive, degenerative, and often fatal. There is no known effective treatment or cure. SCA can affect anyone of any age. The disease is caused by either a recessive or dominant gene. In many cases people are not aware that they carry a relevant gene until they have children who begin to show signs of having the disorder.

Machado–Joseph disease (MJD), also known as Machado–Joseph Azorean disease, Machado's disease, Joseph's disease or spinocerebellar ataxia type 3 (SCA3), is a rare autosomal dominantly inherited neurodegenerative disease that causes progressive cerebellar ataxia, which results in a lack of muscle control and coordination of the upper and lower extremities. The symptoms are caused by a genetic mutation that results in an expansion of abnormal "CAG" trinucleotide repeats in the ATXN3 gene that results in an abnormal form of the protein ataxin which causes degeneration of cells in the hindbrain. Some symptoms, such as clumsiness and rigidity, make MJD commonly mistaken for drunkenness or Parkinson's disease.

Ataxin-1 is a DNA-binding protein which in humans is encoded by the ATXN1 gene.
Ataxin 7 (ATXN7) is a protein of the SCA7 gene, which contains 892 amino acids with an expandable poly(Q) region close to the N-terminus. The expandable poly(Q) motif region in the protein contributes crucially to spinocerebellar ataxia (SCA) pathogenesis by the induction of intranuclear inclusion bodies. ATXN7 is associated with both olivopontocerebellar atrophy type 3 (OPCA3) and spinocerebellar ataxia type 7 (SCA7).
Ataxin is a type of nuclear protein. The class is called ataxin because mutated forms of these proteins and their corresponding genes were found to cause progressive ataxia.
Spinocerebellar ataxia type 6 (SCA6) is a rare, late-onset, autosomal dominant disorder, which, like other types of SCA, is characterized by dysarthria, oculomotor disorders, peripheral neuropathy, and ataxia of the gait, stance, and limbs due to cerebellar dysfunction. Unlike other types, SCA 6 is not fatal. This cerebellar function is permanent and progressive, differentiating it from episodic ataxia type 2 (EA2) where said dysfunction is episodic. In some SCA6 families, some members show these classic signs of SCA6 while others show signs more similar to EA2, suggesting that there is some phenotypic overlap between the two disorders. SCA6 is caused by mutations in CACNA1A, a gene encoding a calcium channel α subunit. These mutations tend to be trinucleotide repeats of CAG, leading to the production of mutant proteins containing stretches of 20 or more consecutive glutamine residues; these proteins have an increased tendency to form intracellular agglomerations. Unlike many other polyglutamine expansion disorders expansion length is not a determining factor for the age that symptoms present.
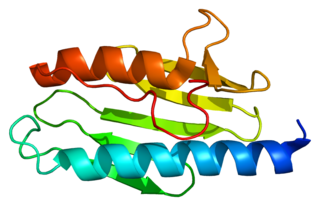
Frataxin is a protein that in humans is encoded by the FXN gene.

Atrophin-1 is a protein that in humans is encoded by the ATN1 gene. The encoded protein includes a serine repeat and a region of alternating acidic and basic amino acids, as well as the variable glutamine repeat. The function of Atrophin-1 has not yet been determined. There is evidence provided by studies of Atrophin-1 in animals to suggest it acts as a transcriptional co-repressor. Atrophin-1 can be found in the nuclear and cytoplasmic compartments of neurons. It is expressed in nervous tissue.

Inositol 1,4,5-trisphosphate receptor type 1 is a protein that in humans is encoded by the ITPR1 gene.
Ataxin-2 is a protein that in humans is encoded by the ATXN2 gene. Mutations in ATXN2 cause spinocerebellar ataxia type 2 (SCA2).

UV excision repair protein RAD23 homolog B is a protein that in humans is encoded by the RAD23B gene.

Cav2.1, also called the P/Q voltage-dependent calcium channel, is a calcium channel found mainly in the brain. Specifically, it is found on the presynaptic terminals of neurons in the brain and cerebellum. Cav2.1 plays an important role in controlling the release of neurotransmitters between neurons. It is composed of multiple subunits, including alpha-1, beta, alpha-2/delta, and gamma subunits. The alpha-1 subunit is the pore-forming subunit, meaning that the calcium ions flow through it. Different kinds of calcium channels have different isoforms (versions) of the alpha-1 subunit. Cav2.1 has the alpha-1A subunit, which is encoded by the CACNA1A gene. Mutations in CACNA1A have been associated with various neurologic disorders, including familial hemiplegic migraine, episodic ataxia type 2, and spinocerebellar ataxia type 6.

Polyglutamine-binding protein 1 (PQBP1) is a protein that in humans is encoded by the PQBP1 gene.

Twinkle protein also known as twinkle mtDNA helicase is a mitochondrial protein that in humans is encoded by the TWNK gene located in the long arm of chromosome 10 (10q24.31).
Sacsin also known as DnaJ homolog subfamily C member 29 (DNAJC29) is a protein that in humans is encoded by the SACS gene. Sacsin is a Hsp70 co-chaperone.
Ataxin 8 opposite strand, also known as ATXN8OS, is a human gene.
Dentatorubral–pallidoluysian atrophy (DRPLA) is an autosomal dominant spinocerebellar degeneration caused by an expansion of a CAG repeat encoding a polyglutamine tract in the atrophin-1 protein. It is also known as Haw River Syndrome and Naito–Oyanagi disease. Although this condition was perhaps first described by Smith et al. in 1958, and several sporadic cases have been reported from Western countries, this disorder seems to be very rare except in Japan.
Autosomal dominant cerebellar ataxia (ADCA) is a form of spinocerebellar ataxia inherited in an autosomal dominant manner. ADCA is a genetically inherited condition that causes deterioration of the nervous system leading to disorder and a decrease or loss of function to regions of the body.
Spinocerebellar ataxia type 1 (SCA1) is a rare autosomal dominant disorder, which, like other spinocerebellar ataxias, is characterized by neurological symptoms including dysarthria, hypermetric saccades, and ataxia of gait and stance. This cerebellar dysfunction is progressive and permanent. First onset of symptoms is normally between 30 and 40 years of age, though juvenile onset can occur. Death typically occurs within 10 to 30 years from onset.

Nancy M. Bonini is an American neuroscientist and geneticist, best known for pioneering the use of Drosophila as a model organism to study neurodegeneration of the human brain. Using the Drosophila model approach, Bonini's laboratory has identified genes and pathways that are important in the development and progression of neurodegenerative diseases such as Amyotrophic lateral sclerosis, Alzheimer's disease, and Parkinson's disease, as well as aging, neural injury and regeneration, and response to environmental toxins.
PDB gallery | |
|---|---|
|


