Protein targeting or protein sorting is the biological mechanism by which proteins are transported to their appropriate destinations within or outside the cell. Proteins can be targeted to the inner space of an organelle, different intracellular membranes, the plasma membrane, or to the exterior of the cell via secretion. Information contained in the protein itself directs this delivery process. Correct sorting is crucial for the cell; errors or dysfunction in sorting have been linked to multiple diseases.

The intermembrane space (IMS) is the space occurring between or involving two or more membranes. In cell biology, it is most commonly described as the region between the inner membrane and the outer membrane of a mitochondrion or a chloroplast. It also refers to the space between the inner and outer nuclear membranes of the nuclear envelope, but is often called the perinuclear space. The IMS of mitochondria plays a crucial role in coordinating a variety of cellular activities, such as regulation of respiration and metabolic functions. Unlike the IMS of the mitochondria, the IMS of the chloroplast does not seem to have any obvious function.

Tafazzin is a protein that in humans is encoded by the TAFAZZIN gene. Tafazzin is highly expressed in cardiac and skeletal muscle, and functions as a phospholipid-lysophospholipid transacylase. It catalyzes remodeling of immature cardiolipin to its mature composition containing a predominance of tetralinoleoyl moieties. Several different isoforms of the tafazzin protein are produced from the TAFAZZIN gene. A long form and a short form of each of these isoforms is produced; the short form lacks a hydrophobic leader sequence and may exist as a cytoplasmic protein rather than being membrane-bound. Other alternatively spliced transcripts have been described but the full-length nature of all these transcripts is not known. Most isoforms are found in all tissues, but some are found only in certain types of cells. Mutations in the TAFAZZIN gene have been associated with mitochondrial deficiency, Barth syndrome, dilated cardiomyopathy (DCM), hypertrophic DCM, endocardial fibroelastosis, left ventricular noncompaction (LVNC), breast cancer, papillary thyroid carcinoma, non-small cell lung cancer, glioma, gastric cancer, thyroid neoplasms, and rectal cancer.

The TIM/TOM complex is a protein complex in cellular biochemistry which translocates proteins produced from nuclear DNA through the mitochondrial membrane for use in oxidative phosphorylation. In enzymology, the complex is described as an mitochondrial protein-transporting ATPase, or more systematically ATP phosphohydrolase , as the TIM part requires ATP hydrolysis to work.

Mitochondrial membrane transport proteins, also known as mitochondrial carrier proteins, are proteins which exist in the membranes of mitochondria. They serve to transport molecules and other factors, such as ions, into or out of the organelles. Mitochondria contain both an inner and outer membrane, separated by the inter-membrane space, or inner boundary membrane. The outer membrane is porous, whereas the inner membrane restricts the movement of all molecules. The two membranes also vary in membrane potential and pH. These factors play a role in the function of mitochondrial membrane transport proteins. There are 53 discovered human mitochondrial membrane transporters, with many others that are known to still need discovered.
Mitochondrial import inner membrane translocase subunit Tim8 A, also known as deafness-dystonia peptide or protein is an enzyme that in humans is encoded by the TIMM8A gene. This translocase has similarity to yeast mitochondrial proteins that are involved in the import of metabolite transporters from the cytoplasm into the mitochondrial inner membrane. The gene is mutated in deafness-dystonia syndrome and it is postulated that MTS/DFN-1 is a mitochondrial disease caused by a defective mitochondrial protein import system.

DnaJ homolog subfamily C member 3 is a protein that in humans is encoded by the DNAJC3 gene.
Sacsin also known as DnaJ homolog subfamily C member 29 (DNAJC29) is a protein that in humans is encoded by the SACS gene. Sacsin is a Hsp70 co-chaperone.

Mitochondrial import inner membrane translocase subunit Tim13 is an enzyme that in humans is encoded by the TIMM13 gene.

The translocase of the outer membrane (TOM) is a complex of proteins found in the outer mitochondrial membrane of the mitochondria. It allows movement of proteins through this barrier and into the intermembrane space of the mitochondrion. Most of the proteins needed for mitochondrial function are encoded by the nucleus of the cell. The outer membrane of the mitochondrion is impermeable to large molecules greater than 5000 Daltons. The TOM works in conjunction with the translocase of the inner membrane (TIM) to translocate proteins into the mitochondrion. Many of the proteins in the TOM complex, such as TOMM22, were first identified in Neurospora crassa and Saccharomyces cerevisiae. Many of the genes encoding these proteins are designated as TOMM (translocase of the outer mitochondrial membrane) complex genes.

FAD-dependent oxidoreductase domain-containing protein 1 (FOXRED1), also known as H17, or FP634 is an enzyme that in humans is encoded by the FOXRED1 gene. FOXRED1 is an oxidoreductase and complex I-specific molecular chaperone involved in the assembly and stabilization of NADH dehydrogenase (ubiquinone) also known as complex I, which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain. Mutations in FOXRED1 have been associated with Leigh syndrome and infantile-onset mitochondrial encephalopathy.

Mitochondrial import inner membrane translocase subunit TIM44 is an enzyme that in humans is encoded by the TIMM44 gene.

Mitochondrial import receptor subunit TOM70 is a protein that in humans is encoded by the TOMM70A gene.

Mitochondrial import inner membrane translocase subunit Tim9 is an enzyme that in humans is encoded by the TIMM9 gene.
Mitochondrial import inner membrane translocase subunit TIM50 is a protein that in humans is encoded by the TIMM50 gene. Tim50 is a subunit of the Tim23 translocase complex in the inner mitochondrial membrane. Mutations in TIMM50 can lead to epilepsy, severe intellectual disability, and 3-methylglutaconic aciduria. TIMM50 expression is increased in breast cancer cells and decreased in hypertrophic hearts.
Putative tyrosine-protein phosphatase auxilin is an enzyme that in humans is encoded by the DNAJC6 gene.
The translocase of the inner membrane (TIM) is a complex of proteins found in the inner mitochondrial membrane of the mitochondria. Components of the TIM complex facilitate the translocation of proteins across the inner membrane and into the mitochondrial matrix. They also facilitate the insertion of proteins into the inner mitochondrial membrane, where they must reside in order to function, these mainly include members of the mitochondrial carrier family of proteins.

Mitochondrial import inner membrane translocase subunit TIM16 also known as presequence translocated-associated motor subunit PAM16, mitochondria-associated granulocyte macrophage CSF-signaling molecule, or presequence translocated-associated motor subunit PAM16 is a protein that in humans is encoded by the PAM16 gene.

DnaJ homolog subfamily C member 30 (DNAJC30), also known as Williams Beuren syndrome chromosome region 18 protein (WBSCR18), is a protein that in humans is encoded by the DNAJC30 gene. This intronless gene encodes a member of the DNAJ molecular chaperone homology domain-containing protein family. This gene is deleted in Williams syndrome, a multisystem developmental disorder caused by the deletion of contiguous genes at 7q11.23.
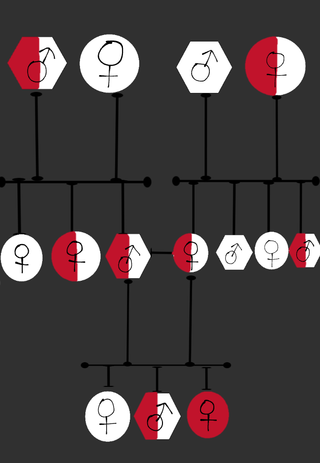
Dilated cardiomyopathy with ataxia syndrome is a multi-systemic hereditary disorder that is characterized by heart abnormalities and problems with coordination, movement and balance.


