
Buprenorphine, sold under the brand name Subutex among others, is an opioid used to treat opioid use disorder, acute pain, and chronic pain. It can be used under the tongue (sublingual), in the cheek (buccal), by injection, as a skin patch (transdermal), or as an implant. For opioid use disorder, the patient must have moderate opioid withdrawal symptoms before buprenorphine can be administered under direct observation of a health-care provider.
Functional selectivity is the ligand-dependent selectivity for certain signal transduction pathways relative to a reference ligand at the same receptor. Functional selectivity can be present when a receptor has several possible signal transduction pathways. To which degree each pathway is activated thus depends on which ligand binds to the receptor. Functional selectivity, or biased signaling, is most extensively characterized at G protein coupled receptors (GPCRs). A number of biased agonists, such as those at muscarinic M2 receptors tested as analgesics or antiproliferative drugs, or those at opioid receptors that mediate pain, show potential at various receptor families to increase beneficial properties while reducing side effects. For example, pre-clinical studies with G protein biased agonists at the μ-opioid receptor show equivalent efficacy for treating pain with reduced risk for addictive potential and respiratory depression. Studies within the chemokine receptor system also suggest that GPCR biased agonism is physiologically relevant. For example, a beta-arrestin biased agonist of the chemokine receptor CXCR3 induced greater chemotaxis of T cells relative to a G protein biased agonist.

The μ-opioid receptors (MOR) are a class of opioid receptors with a high affinity for enkephalins and beta-endorphin, but a low affinity for dynorphins. They are also referred to as μ(mu)-opioid peptide (MOP) receptors. The prototypical μ-opioid receptor agonist is morphine, the primary psychoactive alkaloid in opium and for which the receptor was named, with mu being the first letter of Morpheus, the compound's namesake in the original Greek. It is an inhibitory G-protein coupled receptor that activates the Gi alpha subunit, inhibiting adenylate cyclase activity, lowering cAMP levels.

The nociceptin opioid peptide receptor (NOP), also known as the nociceptin/orphanin FQ (N/OFQ) receptor or kappa-type 3 opioid receptor, is a protein that in humans is encoded by the OPRL1 gene. The nociceptin receptor is a member of the opioid subfamily of G protein-coupled receptors whose natural ligand is the 17 amino acid neuropeptide known as nociceptin (N/OFQ). This receptor is involved in the regulation of numerous brain activities, particularly instinctive and emotional behaviors. Antagonists targeting NOP are under investigation for their role as treatments for depression and Parkinson's disease, whereas NOP agonists have been shown to act as powerful, non-addictive painkillers in non-human primates.

The δ-opioid receptor, also known as delta opioid receptor or simply delta receptor, abbreviated DOR or DOP, is an inhibitory 7-transmembrane G-protein coupled receptor coupled to the G protein Gi/G0 and has enkephalins as its endogenous ligands. The regions of the brain where the δ-opioid receptor is largely expressed vary from species model to species model. In humans, the δ-opioid receptor is most heavily expressed in the basal ganglia and neocortical regions of the brain.
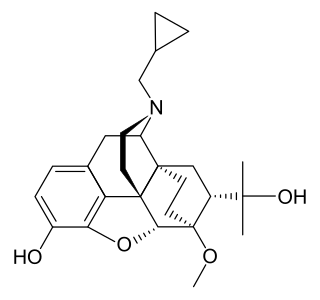
Diprenorphine, also known as diprenorfin, is a non-selective, high-affinity, weak partial agonist of the μ- (MOR), κ- (KOR), and δ-opioid receptor (DOR) which is used in veterinary medicine as an opioid antagonist. It is used to reverse the effects of super-potent opioid analgesics such as etorphine and carfentanil that are used for tranquilizing large animals. The drug is not approved for use in humans.
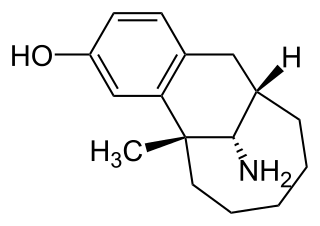
Dezocine, sold under the brand name Dalgan, is an atypical opioid analgesic which is used in the treatment of pain. It is used by intravenous infusion and intramuscular injection.

SNC-80 is an opioid analgesic compound that selectively activates μ–δ opioid receptor heteromers and is used primarily in scientific research. Discovered in 1994, SNC-80 was a pioneering non-peptide compound regarded as a highly selective agonist for the δ-opioid receptor.

RB-101 is a drug that acts as an enkephalinase inhibitor, which is used in scientific research.

(+)-BW373U86 is an opioid analgesic drug used in scientific research.

DPI-287 is an opioid drug that is used in scientific research. It is a highly selective agonist for the δ-opioid receptor, which produces less convulsions than most drugs from this family. It has antidepressant-like effects.

SC-17599 is a steroid derivative drug discovered in 1968 which acts as a selective μ-opioid receptor agonist, with little or no affinity for the δ-opioid or κ-opioid receptors. It is an active analgesic in vivo, more potent than codeine or pethidine but slightly less potent than morphine, and produces similar effects to morphine in animals but with less sedation

Oxymorphazone is an opioid analgesic drug related to oxymorphone. Oxymorphazone is a potent and long acting μ-opioid agonist which binds irreversibly to the receptor, forming a covalent bond which prevents it from detaching once bound. This gives it an unusual pharmacological profile, and while oxymorphazone is only around half the potency of oxymorphone, with higher doses the analgesic effect becomes extremely long lasting, with a duration of up to 48 hours. However, tolerance to analgesia develops rapidly with repeated doses, as chronically activated opioid receptors are rapidly internalised by β-arrestins, similar to the results of non-covalent binding by repeated doses of agonists with extremely high binding affinity such as lofentanil.

β-Chlornaltrexamine (β-CNA) is a non-selective irreversible antagonist of the μ-opioid receptor (MOR), the δ-opioid receptor (DOR), and the κ-opioid receptor (KOR), which forms a covalent bond to the binding sites of these receptors and has ultra-long-lasting opioid antagonist effects. Although it is predominantly antagonistic, β-CNA also shows some irreversible mixed agonist–antagonist activity at the MOR and KOR and some associated analgesic effects. Its alkylating group is a bis(chloroalkyl)amino-residue similar to that of the nitrogen mustards.

GR-89696 is a drug which acts as a highly selective κ-opioid agonist. It has been studied in various animal species, and has been described as selective for the κ2 subtype. Recent studies have suggested that GR-89696 and related κ2-selective agonists may be useful for preventing the itching which is a common side effect of conventional opioid analgesic drugs, without the additional side effects of non-selective kappa agonists. The structure bound to the κ-opioid receptor has been reported.

Clocinnamox is a selective and irreversible antagonist of the μ-opioid receptor. Closely related compounds include methocinnamox (MCAM) and methoclocinnamox (MCCAM). They were derived via structural modification of buprenorphine. Clocinnamox was first described in the scientific literature by 1992.

Buprenorphine/samidorphan is a combination formulation of buprenorphine and samidorphan which is under development as an add on to antidepressants in treatment-resistant depression (TRD).
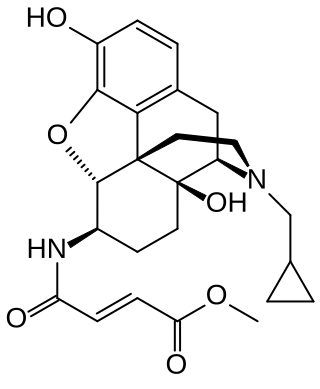
β-Funaltrexamine (β-FNA) is an irreversible opioid antagonist that was used to create the first crystal structure of the μ-opioid receptor (MOR). It is selective for antagonism of the MOR over the δ-opioid receptor (DOR) and κ-opioid receptor (KOR). Chemically, it is a naltrexone derivative with a methyl-fumaramide group in the 6-position. In addition to its MOR irreversible antagonism, β-FNA is a reversible agonist of the κ-opioid receptor (KOR) and produces KOR-mediated analgesic effects in animals. This has limited its usefulness and contributed to the development of methocinnamox as a more selective functionally irreversible antagonist of the MOR with no significant opioid agonistic actions.
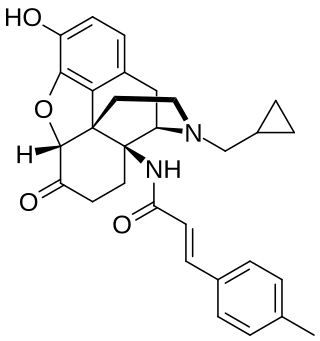
Methocinnamox (MCAM) is an opioid receptor antagonist. It is a pseudo-irreversible non-competitive antagonist of the μ-opioid receptor and a competitive antagonist of the κ- and δ-opioid receptors. The drug has a very long duration of action of up to months with a single dose due to its pseudo-irreversibility. It is administered in animals by intravenous or subcutaneous injection.

Methoclocinnamox is a selective pseudo-irreversible partial agonist of the μ-opioid receptor (MOR). It shows a mixture of opioid agonist- and antagonist-like effects. The drug has long-lasting effects and is insurmountable by other MOR ligands.
