
Norbuprenorphine is a major active metabolite of the opioid modulator buprenorphine. It is a μ-opioid, δ-opioid, and nociceptin receptor full agonist, and a κ-opioid receptor partial agonist. In rats, unlike buprenorphine, norbuprenorphine produces marked respiratory depression but with very little antinociceptive effect. In explanation of these properties, norbuprenorphine has been found to be a high affinity P-glycoprotein substrate, and in accordance, shows very limited blood-brain-barrier penetration.

The nociceptin opioid peptide receptor (NOP), also known as the nociceptin/orphanin FQ (N/OFQ) receptor or kappa-type 3 opioid receptor, is a protein that in humans is encoded by the OPRL1 gene. The nociceptin receptor is a member of the opioid subfamily of G protein-coupled receptors whose natural ligand is the 17 amino acid neuropeptide known as nociceptin (N/OFQ). This receptor is involved in the regulation of numerous brain activities, particularly instinctive and emotional behaviors. Antagonists targeting NOP are under investigation for their role as treatments for depression and Parkinson's disease, whereas NOP agonists have been shown to act as powerful, non-addictive painkillers in non-human primates.

The δ-opioid receptor, also known as delta opioid receptor or simply delta receptor, abbreviated DOR or DOP, is an inhibitory 7-transmembrane G-protein coupled receptor coupled to the G protein Gi/G0 and has enkephalins as its endogenous ligands. The regions of the brain where the δ-opioid receptor is largely expressed vary from species model to species model. In humans, the δ-opioid receptor is most heavily expressed in the basal ganglia and neocortical regions of the brain.

SNC-80 is an opioid analgesic compound that selectively activates μ–δ opioid receptor heteromers and is used primarily in scientific research. Discovered in 1994, SNC-80 was a pioneering non-peptide compound regarded as a highly selective agonist for the δ-opioid receptor.

(+)-BW373U86 is an opioid analgesic drug used in scientific research.
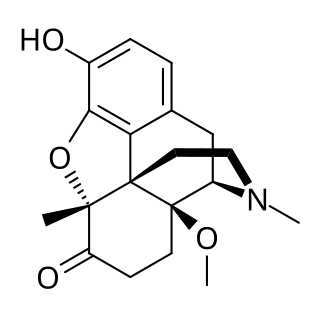
14-Methoxymetopon is an experimental opioid drug developed by a team led by Professor Helmut Schmidhammer at the University of Innsbruck in the mid-1990s. It is a derivative of metopon in which a methoxy group has been inserted at the 14-position. It is a highly potent analgesic drug that is around 500 times stronger than morphine when administered systemically; however, when given spinally or supraspinally, it exhibits analgesic activity up to a million fold greater than morphine. It binds strongly to the μ-opioid receptor and activates it to a greater extent than most similar opioid drugs. This produces an unusual pharmacological profile, and although 14-methoxymetopon acts as a potent μ-opioid full agonist in regard to some effects such as analgesia, a ceiling effect is seen on other effects such as constipation and respiratory depression which is believed to involve interaction with the κ-opioid receptor

DPI-221 is an opioid drug that is used in scientific research. It is a highly selective agonist for the δ-opioid receptor, which produces fewer convulsions than most drugs from this family.

DPI-287 is an opioid drug that is used in scientific research. It is a highly selective agonist for the δ-opioid receptor, which produces less convulsions than most drugs from this family. It has antidepressant-like effects.

RWJ-394674 is a drug that is used in scientific research. It is a potent, orally active analgesic drug that produces little respiratory depression. RWJ-394674 itself is a potent and selective agonist for δ-opioid receptors, with a Ki of 0.24 nM at δ and 72 nM at μ. However once inside the body, RWJ-394674 is dealkylated to its monodesethyl metabolite RWJ-413216, which is a potent agonist at the μ-opioid receptor and has less affinity for δ. The effect of RWJ-394674 when administered in vivo thus produces potent agonist effects at both μ and δ receptors through the combined actions of the parent drug and its active metabolite, with the δ-agonist effects counteracting the respiratory depression from the μ-opioid effects, and the only prominent side-effect being sedation.

SC-17599 is a steroid derivative drug discovered in 1968 which acts as a selective μ-opioid receptor agonist, with little or no affinity for the δ-opioid or κ-opioid receptors. It is an active analgesic in vivo, more potent than codeine or pethidine but slightly less potent than morphine, and produces similar effects to morphine in animals but with less sedation

IBNtxA, or 3-iodobenzoyl naltrexamine, is an atypical opioid analgesic drug derived from naltrexone. In animal studies it produces potent analgesic effects that are blocked by levallorphan and so appear to be μ-opioid mediated, but it fails to produce constipation or respiratory depression, and is neither rewarding or aversive in conditioned place preference protocols. These unusual properties are thought to result from agonist action at a splice variant or heterodimer of the μ-opioid receptor, rather than at the classical full length form targeted by conventional opioid drugs.

Biphalin is a dimeric enkephalin endogenous peptide (Tyr-D-Ala-Gly-Phe-NH)2 composed of two tetrapeptides derived from enkephalins, connected 'tail-to-tail' by a hydrazide bridge. The presence of two distinct pharmacophores confers on biphalin a high affinity for both μ and δ opioid receptors (with an EC50 of about 1–5 nM for both μ and δ receptors), therefore it has analgesic activity. Biphalin presents a considerable antinociceptive profile. In fact, when administered intracerebroventricularly in mice, biphalin displays a potency almost 7-fold greater than that of the ultra-potent alkaloid agonist, etorphine and 7000-fold greater than morphine; biphalin and morphine were found to be equipotent after intraperitoneal administration. The extraordinary in vivo potency shown by this compound is coupled with low side-effects, in particular, to produce no dependency in chronic use. For these reasons, several efforts have been carried out in order to obtain more information about structure-activity relationship (SAR). Results clearly indicate that, at least for μ receptor binding, the presence of two pharmacophores is not necessary; Tyr1 is indispensable for analgesic activity, while replacing Phe at the position 4 and 4' with non-aromatic, but lipophilic amino acids does not greatly change the binding properties and in general 4,4' positions are found to be important to design biphalin analogues with increased potency and modified μ/δ selectivity. The hydrazide linker is not fundamental for activity or binding, and it can be conveniently substituted by different conformationally constrained cycloaliphatic diamine linkers.
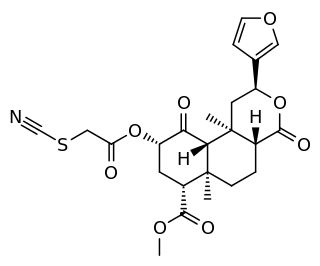
RB-64 is a semi-synthetic derivative of salvinorin A. It is an irreversible agonist, with a reactive thiocyanate group that forms a bond to the κ-opioid receptor (KOR), resulting in very high potency. It is functionally selective, activating G proteins more potently than β-arrestin-2. RB-64 has a bias factor of up to 96 and is analgesic with fewer of the side-effects associated with unbiased KOR agonists. The analgesia is long-lasting. Compared with unbiased agonists, RB-64 evokes considerably less receptor internalization.

Cebranopadol is an opioid analgesic of the benzenoid class which is currently under development internationally by Grünenthal, a German pharmaceutical company, and its partner Depomed, a pharmaceutical company in the United States, for the treatment of a variety of different acute and chronic pain states. As of November 2014, it is in phase III clinical trials.

Thienorphine is a very potent, extremely long-acting, orally-active opioid analgesic with mixed agonist–antagonist properties which was developed by the Beijing Institute of Pharmacology and Toxicology as a potential treatment for opioid dependence. It is a high-affinity, balanced ligand of the μ-, δ-, and κ-opioid receptors, behaving as a partial agonist of the μ- and κ-opioid receptors and as an antagonist of the δ-opioid receptor. It also possesses relatively low affinity for the nociceptin receptor, where it acts as an antagonist.

PZM21 is an experimental opioid analgesic drug that is being researched for the treatment of pain. It is claimed to be a functionally selective μ-opioid receptor agonist which produces μ-opioid receptor mediated G protein signaling, with potency and efficacy similar to morphine, but with less β-arrestin 2 recruitment. However, recent reports highlight that this might be due to its low intrinsic efficacy, rather than functional selectivity or 'G protein bias' as initially reported. In tests on mice, PZM21 was slightly less potent than morphine or TRV130 as an analgesic, but also had significantly reduced adverse effects, with less constipation than morphine, and very little respiratory depression, even at high doses. This research was described as a compelling example of how modern high-throughput screening techniques can be used to discover new chemotypes with specific activity profiles, even at targets such as the μ-opioid receptor which have already been thoroughly investigated. More recent research has suggested however that at higher doses, PZM21 is capable of producing classic opioid side effects such as respiratory depression and development of tolerance and may have only limited functional selectivity.
Peripherally acting μ-opioid receptor antagonists (PAMORAs) are a class of chemical compounds that are used to reverse adverse effects caused by opioids interacting with receptors outside the central nervous system (CNS), mainly those located in the gastrointestinal tract. PAMORAs are designed to specifically inhibit certain opioid receptors in the gastrointestinal tract and with limited ability to cross the blood–brain barrier. Therefore, PAMORAs do not affect the analgesic effects of opioids within the central nervous system.
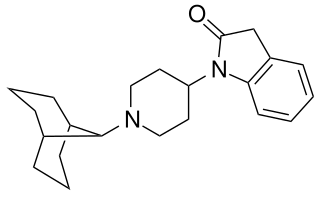
SR-16435 is a drug which acts as a potent partial agonist at both the μ-opioid receptor and nociceptin receptor. In animal studies it was found to be a potent analgesic, with results suggestive of reduced development of tolerance and increased activity against neuropathic pain compared to classic μ-selective agonists.
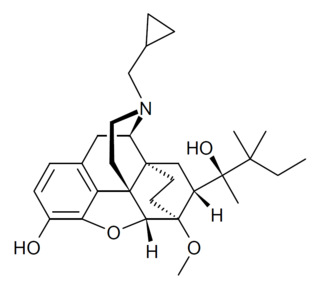
BU08028 is a drug which acts as an extremely potent partial agonist at both the μ-opioid receptor and nociceptin receptor. It is a homologue of buprenorphine extended by just one carbon on the side chain, but has relatively greater activity at the nociceptin receptor, which is thought to reduce the abuse potential without compromising analgesia.
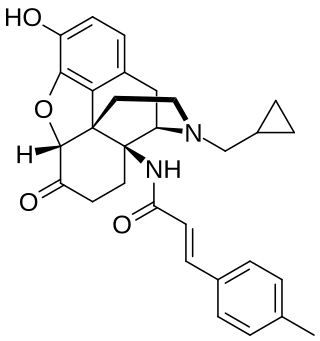
Methocinnamox (MCAM) is an opioid receptor antagonist. It is a pseudo-irreversible non-competitive antagonist of the μ-opioid receptor and a competitive antagonist of the κ- and δ-opioid receptors. The drug has a very long duration of action of up to months with a single dose due to its pseudo-irreversibility. It is administered in animals by intravenous or subcutaneous injection.
