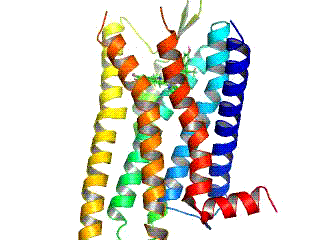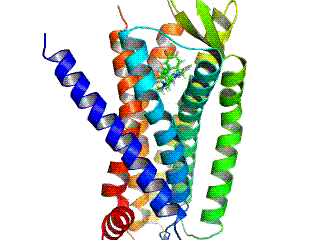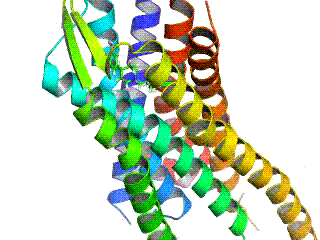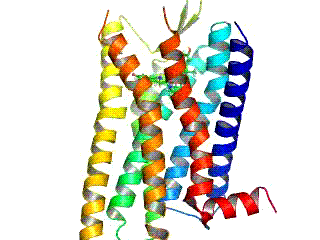
Opioid receptors are a group of inhibitory G protein-coupled receptors with opioids as ligands. The endogenous opioids are dynorphins, enkephalins, endorphins, endomorphins and nociceptin. The opioid receptors are ~40% identical to somatostatin receptors (SSTRs). Opioid receptors are distributed widely in the brain, in the spinal cord, on peripheral neurons, and digestive tract.

An opioid antagonist, or opioid receptor antagonist, is a receptor antagonist that acts on one or more of the opioid receptors.

The μ-opioid receptors (MOR) are a class of opioid receptors with a high affinity for enkephalins and beta-endorphin, but a low affinity for dynorphins. They are also referred to as μ(mu)-opioid peptide (MOP) receptors. The prototypical μ-opioid receptor agonist is morphine, the primary psychoactive alkaloid in opium and for which the receptor was named, with mu being the first letter of Morpheus, the compound's namesake in the original Greek. It is an inhibitory G-protein coupled receptor that activates the Gi alpha subunit, inhibiting adenylate cyclase activity, lowering cAMP levels.

The nociceptin opioid peptide receptor (NOP), also known as the nociceptin/orphanin FQ (N/OFQ) receptor or kappa-type 3 opioid receptor, is a protein that in humans is encoded by the OPRL1 gene. The nociceptin receptor is a member of the opioid subfamily of G protein-coupled receptors whose natural ligand is the 17 amino acid neuropeptide known as nociceptin (N/OFQ). This receptor is involved in the regulation of numerous brain activities, particularly instinctive and emotional behaviors. Antagonists targeting NOP are under investigation for their role as treatments for depression and Parkinson's disease, whereas NOP agonists have been shown to act as powerful, non-addictive painkillers in non-human primates.

The δ-opioid receptor, also known as delta opioid receptor or simply delta receptor, abbreviated DOR or DOP, is an inhibitory 7-transmembrane G-protein coupled receptor coupled to the G protein Gi/G0 and has enkephalins as its endogenous ligands. The regions of the brain where the δ-opioid receptor is largely expressed vary from species model to species model. In humans, the δ-opioid receptor is most heavily expressed in the basal ganglia and neocortical regions of the brain.
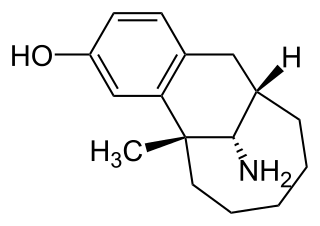
Dezocine, sold under the brand name Dalgan, is an atypical opioid analgesic which is used in the treatment of pain. It is used by intravenous infusion and intramuscular injection.

Proglumide (Milid) is a drug that inhibits gastrointestinal motility and reduces gastric secretions. It acts as a cholecystokinin antagonist, which blocks both the CCKA and CCKB subtypes. It was used mainly in the treatment of stomach ulcers, although it has now been largely replaced by newer drugs for this application.
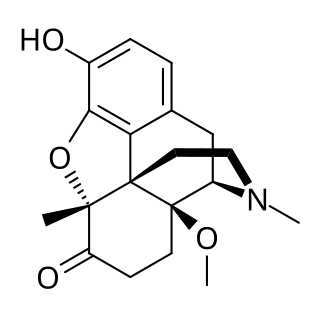
14-Methoxymetopon is an experimental opioid drug developed by a team led by Professor Helmut Schmidhammer at the University of Innsbruck in the mid-1990s. It is a derivative of metopon in which a methoxy group has been inserted at the 14-position. It is a highly potent analgesic drug that is around 500 times stronger than morphine when administered systemically; however, when given spinally or supraspinally, it exhibits analgesic activity up to a million fold greater than morphine. It binds strongly to the μ-opioid receptor and activates it to a greater extent than most similar opioid drugs. This produces an unusual pharmacological profile, and although 14-methoxymetopon acts as a potent μ-opioid full agonist in regard to some effects such as analgesia, a ceiling effect is seen on other effects such as constipation and respiratory depression which is believed to involve interaction with the κ-opioid receptor
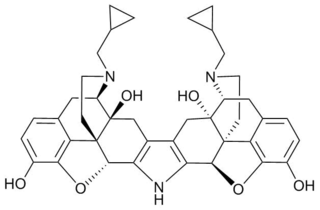
Norbinaltorphimine is an opioid antagonist used in scientific research. It is one of the few opioid antagonists available that is highly selective for the κ-opioid receptor, and blocks this receptor without affecting the μ- or δ-opioid receptors, although it is less selective in vivo than in isolated tissues. nor-BNI blocks the effects of κ-opioid agonists in animal models, and produces antidepressant and anxiolytic-like effects.
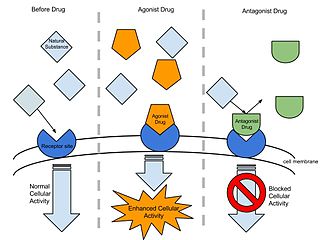
In pharmacology the term agonist-antagonist or mixed agonist/antagonist is used to refer to a drug which under some conditions behaves as an agonist while under other conditions, behaves as an antagonist.
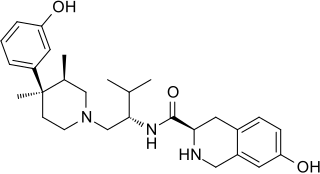
JDTic is a selective, long-acting ("inactivating") antagonist of the κ-opioid receptor (KOR). JDTic is a 4-phenylpiperidine derivative, distantly related structurally to analgesics such as pethidine and ketobemidone, and more closely to the MOR antagonist alvimopan. In addition, it is structurally distinct from other KOR antagonists such as norbinaltorphimine. JDTic has been used to create crystal structures of KOR [ PDB: 4DJH, 6VI4].

Alazocine, also known more commonly as N-allylnormetazocine (NANM), is a synthetic opioid analgesic of the benzomorphan family related to metazocine, which was never marketed. In addition to its opioid activity, the drug is a sigma receptor agonist, and has been used widely in scientific research in studies of this receptor. Alazocine is described as a potent analgesic, psychotomimetic or hallucinogen, and opioid antagonist. Moreover, one of its enantiomers was the first compound that was found to selectively label the σ1 receptor, and led to the discovery and characterization of the receptor.

8-Carboxamidocyclazocine (8-CAC) is an opioid analgesic drug related to cyclazocine, discovered by medicinal chemist Mark P. Wentland and co-workers in Cogswell Laboratory at Rensselaer Polytechnic Institute. Similarly to cyclazocine, 8-CAC acts as an agonist at both the μ- and κ-opioid receptors, but has a much longer duration of action than cyclazocine, and does not have μ antagonist activity. Unexpectedly, it was discovered that the phenolic hydroxyl group of cyclazocine could be replaced by a carboxamido group with only slight loss of potency at opioid receptors, and this discovery has subsequently been used to develop many novel opioid derivatives where the phenolic hydroxy group has been replaced by either carboxamide or a variety of larger groups. Due to their strong κ-opioid agonist activity, these drugs are not suited for use as analgesics in humans, but have instead been researched as potential drugs for the treatment of cocaine addiction.

Samidorphan is an opioid antagonist that in the form of olanzapine/samidorphan is used in the treatment of schizophrenia and bipolar disorder. Samidorphan reduces the weight gain associated with olanzapine. Samidorphan is taken by mouth.

Buprenorphine/samidorphan is a combination formulation of buprenorphine and samidorphan which is under development as an add on to antidepressants in treatment-resistant depression (TRD).

Aticaprant, also known by its developmental codes JNJ-67953964, CERC-501, and LY-2456302, is a κ-opioid receptor (KOR) antagonist which is under development for the treatment of major depressive disorder. A regulatory application for approval of the medication is expected to be submitted by 2025. Aticaprant is taken by mouth.

6β-Naltrexol, or 6β-hydroxynaltrexone, is a peripherally-selective opioid receptor antagonist related to naltrexone. It is a major active metabolite of naltrexone formed by hepatic dihydrodiol dehydrogenase enzymes. With naltrexone therapy, 6β-naltrexol is present at approximately 10- to 30-fold higher concentrations than naltrexone at steady state due to extensive first-pass metabolism of naltrexone into 6β-naltrexol. In addition to being an active metabolite of naltrexone, 6β-naltrexol was itself studied for the treatment of opioid-induced constipation. It was found to be effective and well-tolerated, and did not precipitate opioid withdrawal symptoms or interfere with opioid pain relief, but development was not further pursued.

AT-076 is a so-called opioid "pan" antagonist and is the first reasonably balanced antagonist known of all four opioid receptor types. It acts as a silent antagonist of all four of the opioid receptors, behaving as a competitive antagonist of the μ-opioid receptor and δ-opioid receptor and as a noncompetitive antagonist of the κ-opioid receptor and nociceptin receptor. AT-076 was derived from the selective κ-opioid receptor antagonist JDTic via removal of the 3,4-dimethyl group of the trans-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine antagonist scaffold which increased affinity for the nociceptin receptor by 10-fold and for the μ- and δ-opioid receptors by 3-6-fold.
Peripherally acting μ-opioid receptor antagonists (PAMORAs) are a class of chemical compounds that are used to reverse adverse effects caused by opioids interacting with receptors outside the central nervous system (CNS), mainly those located in the gastrointestinal tract. PAMORAs are designed to specifically inhibit certain opioid receptors in the gastrointestinal tract and with limited ability to cross the blood–brain barrier. Therefore, PAMORAs do not affect the analgesic effects of opioids within the central nervous system.

HS665 is a drug which acts as a potent and selective κ-opioid receptor agonist, and has analgesic effects in animal studies. HS665 is not an agonist for the mu receptor, leading to less potential for abuse.

