Protactinium is a chemical element; it has symbolPa and atomic number 91. It is a dense, radioactive, silvery-gray actinide metal which readily reacts with oxygen, water vapor, and inorganic acids. It forms various chemical compounds, in which protactinium is usually present in the oxidation state +5, but it can also assume +4 and even +3 or +2 states. Concentrations of protactinium in the Earth's crust are typically a few parts per trillion, but may reach up to a few parts per million in some uraninite ore deposits. Because of its scarcity, high radioactivity, and high toxicity, there are currently no uses for protactinium outside scientific research, and for this purpose, protactinium is mostly extracted from spent nuclear fuel.
The element was first identified in 1913 by Kazimierz Fajans and Oswald Helmuth Göhring and named "brevium" because of the short half-life of the specific isotope studied, 234mPa. A more stable isotope of protactinium, 231Pa, was discovered in 1917/18 by Lise Meitner in collaboration with Otto Hahn, and they named the element protactinium.[9] In 1949, the IUPAC chose the name "protactinium" and confirmed Hahn and Meitner as its discoverers. The new name meant "(nuclear) precursor of actinium,"[10] suggesting that actinium is a product of radioactive decay of protactinium. John Arnold Cranston (working with Frederick Soddy and Ada Hitchins) is also credited with discovering the most stable isotope in 1915, but he delayed his announcement due to being called for service in the First World War.[11]
The longest-lived and most abundant (nearly 100%) naturally occurring isotope of protactinium, 231Pa, has a half-life of 32,760 years and occurs in the decay chain of uranium-235. Much smaller trace amounts of the short-lived 234Pa and its nuclear isomer234mPa occur in the decay chain of uranium-238. 233Pa occurs as a result of the decay of thorium-233 as part of the chain of events necessary to produce uranium-233 by neutron irradiation of 232Th. It is an undesired intermediate product in thorium-basednuclear reactors, and is therefore removed from the active zone of the reactor during the breeding process. Ocean science uses the element to understand the ancient ocean's geography: analysis of the relative concentrations of various uranium, thorium, and protactinium isotopes in water and minerals is used in radiometric dating of sediments up to 175,000 years old, and in modeling of various geological processes.[12]
Dmitri Mendeleev's 1871 periodic table with a gap for protactinium on the bottom row of the chart, between thorium and uranium
In 1871, Dmitri Mendeleevpredicted the existence of an element between thorium and uranium.[13] The actinide series was unknown at the time, so Mendeleev positioned uranium below tungsten in group VI, and thorium below zirconium in group IV, leaving the space below tantalum in group V empty. Until the general acceptance of the actinide concept in the late 1940s, periodic tables were published with this structure.[14] For a long time, chemists searched for eka-tantalum[note 1] as an element with similar chemical properties to tantalum, making a discovery of protactinium nearly impossible. Tantalum's heavier analogue was later found to be the transuranic element dubnium – although dubnium is more chemically similar to protactinium, not tantalum.[15]
In 1900, William Crookes isolated protactinium as an intensely radioactive material from uranium; however, he could not characterize it as a new chemical element and thus named it uranium X (UX).[13][16][17] Crookes dissolved uranium nitrate in ether, and the residual aqueous phase contained most of the 234 90Th and 234 91Pa. His method was used into the 1950s to isolate 234 90Th and 234 91Pa from uranium compounds.[18] Protactinium was first identified in 1913, when Kasimir Fajans and Oswald Helmuth Göhring encountered the isotope 234mPa during their studies of the decay chains of uranium-238: 238 92U → 234 90Th → 234m 91Pa → 234 92U. They named the new element "brevium" (from the Latin word brevis, meaning brief or short) because of the short half-life of 1.16 minutes for 234m 91Pa (uraniumX2).[19][20][21][22][23][24] In 1917–18, two groups of scientists, Lise Meitner in collaboration with Otto Hahn of Germany and Frederick Soddy and John Cranston of Great Britain, independently discovered another isotope, 231Pa, having a much longer half-life of 32,760 years.[9][23][25] Meitner changed the name "brevium" to protactinium as the new element was part of the decay chain of uranium-235 as the parent of actinium (from the Greek: πρῶτοςprôtos, meaning "first, before").[26] The IUPAC confirmed this naming in 1949.[27][28] The discovery of protactinium completed one of the last gaps in early versions of the periodic table, and brought fame to the involved scientists.[29]
Aristid von Grosse produced 2milligrams of Pa2O5 in 1927,[30] and in 1934 first isolated elemental protactinium from 0.1milligrams of Pa2O5.[31] He used two different procedures: in the first, protactinium oxide was irradiated by 35keV electrons in vacuum. In the other, called the van Arkel–de Boer process, the oxide was chemically converted to a halide (chloride, bromide or iodide) and then reduced in a vacuum with an electrically heated metallic filament:[27][32]
2 PaI5 → 2 Pa + 5 I2
In 1961, the United Kingdom Atomic Energy Authority (UKAEA) produced 127grams of 99.9% pure protactinium-231 by processing 60tonnes of waste material in a 12-stage process, at a cost of about US$500,000.[27][33] For many years, this was the world's only significant supply of protactinium, which was provided to various laboratories for scientific studies.[13] The Oak Ridge National Laboratory in the US provided protactinium at a cost of about US$280/gram.[34]
Thirty radioisotopes of protactinium have been discovered, ranging from 210Pa to 239Pa.[35][8] The most stable are 231Pa with a half-life of 32,650 years, 233Pa with a half-life of 26.975 days, and 230Pa with a half-life of 17.4 days. All other isotopes have half-lives shorter than 1.6 days, and the majority of these have half-lives less than 1.8 seconds. Protactinium also has six nuclear isomers, with the most stable being 234mPa (half-life 1.159 minutes).[8]
The longest-lived and most abundant isotope, 231Pa, can fission from fast neutrons exceeding ~1MeV.[36]233Pa, the other isotope of protactinium produced in nuclear reactors, also has a fission threshold of 1MeV.[37]
Occurrence
Protactinium is one of the rarest and most expensive naturally occurring elements. It is found in the form of two isotopes, 231Pa and 234Pa, with the isotope 234Pa occurring in two different energy states. Nearly all natural protactinium is 231Pa. It is an alpha emitter and is formed by the decay of uranium-235, whereas the beta-radiating234Pa is produced as a result of uranium-238 decay. Nearly all uranium-238 (99.8%) decays first to the shorter-lived 234mPa isomer.[38]
Protactinium occurs in uraninite (pitchblende) at concentrations of about 0.3–3 parts231Pa per million parts (ppm) of ore.[13] Whereas the usual content is closer to 0.3ppm[39] (e.g. in Jáchymov, Czech Republic[40]), some ores from the Democratic Republic of the Congo have about 3ppm.[27] Protactinium is homogeneously dispersed in most natural materials and in water, but at much lower concentrations on the order of one part per trillion, corresponding to a radioactivity of 0.1 picocuries (pCi)/g. There is about 500 times more protactinium in sandy soil particles than in water, even when compared to water present in the same sample of soil. Much higher ratios of 2,000 and above are measured in loam soils and clays, such as bentonite.[38][41]
In nuclear reactors
Two major protactinium isotopes, 231Pa and 233Pa, are produced from thorium in nuclear reactors; both are undesirable and are usually removed, thereby adding complexity to the reactor design and operation. In particular, 232Th, via (n, 2n) reactions, produces 231Th, which quickly decays to 231Pa (half-life 25.5 hours). The last isotope, while not a transuranic waste, has a long half-life of 32,760 years, and is a major contributor to the long-term radiotoxicity of spent nuclear fuel.[42]
Protactinium-233 is formed upon neutron capture by 232Th. It either further decays to 233U, or captures another neutron and converts into the non-fissile 234U.[43]233Pa has a relatively long half-life of 27 days and high cross section for neutron capture (the so-called "neutron poison"). Thus, instead of rapidly decaying to the useful 233U, a significant fraction of 233Pa converts to non-fissile isotopes and consumes neutrons, degrading reactor efficiency. To limit the loss of neutrons, 233Pa is extracted from the active zone of thorium molten salt reactors during their operation, so that it can only decay into 233U. Extraction of 233Pa is achieved using columns of molten bismuth with lithium dissolved in it. In short, lithium selectively reduces protactinium salts to protactinium metal, which is then extracted from the molten-salt cycle, while the molten bismuth is merely a carrier, selected due to its low melting point of 271°C, low vapor pressure, good solubility for lithium and actinides, and immiscibility with molten halides.[42]
Before the advent of nuclear reactors, protactinium was separated for scientific experiments from uranium ores. Since reactors have become more common, it is mostly produced as an intermediate product of neutron capture on thorium, used for the production of the fissile 233U:
The isotope 231Pa can be prepared by irradiating 230Th with slow neutrons, converting it to the beta-decaying 231Th; or, by irradiating 232Th with fast neutrons, generating (as one product) 231Th and 2 neutrons.
Protactinium is an actinide positioned in the periodic table to the left of uranium and to the right of thorium, and many of its physical properties are intermediate between its neighboring actinides. Protactinium is denser and more rigid than thorium, but is lighter than uranium; its melting point is lower than that of thorium, but higher than that of uranium. The thermal expansion, electrical, and thermal conductivities of these three elements are comparable and are typical of post-transition metals. The estimated shear modulus of protactinium is similar to that of titanium.[47] Protactinium is a metal with silvery-gray luster that is preserved for some time in air.[27][33] Protactinium easily reacts with oxygen, water vapor, and acids, but not with alkalis.[13]
At room temperature, protactinium crystallizes in the body-centered tetragonal structure, which can be regarded as distorted body-centered cubic lattice; this structure does not change upon compression up to 53 GPa. The structure changes to face-centered cubic (fcc) upon cooling from high temperature, at about 1200°C.[44][48] The thermal expansion coefficient of the tetragonal phase between room temperature and 700°C is 9.9×10−6/°C.[44]
Protactinium is paramagnetic and no magnetic transitions are known for it at any temperature.[49] It becomes superconductive at temperatures below 1.4K.[13][45] Protactinium tetrachloride is paramagnetic at room temperature, but becomes ferromagnetic when cooled to 182K.[50]
Protactinium exists in two major oxidation states: +4 and +5, both in solids and solutions; and the +3 and +2 states, which have been observed in some solids. As the electron configuration of the neutral atom is [Rn]5f26d17s2, the +5 oxidation state corresponds to the low-energy (and thus favored) 5f0 configuration. Both +4 and +5 states easily form hydroxides in water, with the predominant ions being Pa(OH)3+, Pa(OH)2+2, Pa(OH)+3, and Pa(OH)4, all of which are colorless.[51] Other known protactinium ions include PaOF2+ , PaOF+ 2, PaF− 6, PaF2− 7, PaF3− 8, PaOSO+ 4, PaO(SO 4)− 2, PaO(SO 4)3− 3, PaOCl2− 5, and PaCl− 6.[52][53][54][55]
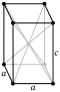
Here, a, b, and c are lattice constants in picometers, No is the space group number, and Z is the number of formula units per unit cell; fcc stands for the face-centered cubic symmetry. Density was not measured directly but calculated from the lattice parameters.
Oxides and oxygen-containing salts
Protactinium oxides are known for the metal oxidation states +2, +4, and +5. The most stable is the white pentoxide Pa2O5, which can be produced by igniting protactinium(V) hydroxide in air at a temperature of 500°C.[63] Its crystal structure is cubic, and the chemical composition is often non-stoichiometric, described as PaO2.25. Another phase of this oxide with orthorhombic symmetry has also been reported.[46][64] The black dioxide PaO2 is obtained from the pentoxide by reducing it at 1550°C with hydrogen. It is not readily soluble in either dilute or concentrated nitric, hydrochloric, or sulfuric acid, but easily dissolves in hydrofluoric acid.[46] The dioxide can be converted back to pentoxide by heating in oxygen-containing atmosphere to 1100°C.[64] The monoxide PaO has only been observed as a thin coating on protactinium metal, but not in an isolated bulk form.[46]
Protactinium forms mixed binary oxides with various metals. With alkali metals A, the crystals have a chemical formula APaO3 and perovskite structure; A3PaO4 and distorted rock-salt structure; or A7PaO6, where oxygen atoms form a hexagonal close-packed lattice. In all of these materials, the protactinium ions are octahedrally coordinated.[65][66] The pentoxide Pa2O5 combines with rare-earth metal oxides R2O3 to form various nonstoichiometric mixed-oxides, also of perovskite structure.[67]
Protactinium oxides are basic; they easily convert to hydroxides and can form various salts, such as sulfates, phosphates, nitrates, etc. The nitrate is usually white but can be brown due to radiolytic decomposition. Heating the nitrate in air at 400°C converts it to the white protactinium pentoxide.[68] The polytrioxophosphate Pa(PO3)4 can be produced by reacting the difluoride sulfate PaF2SO4 with phosphoric acid (H3PO4) under an inert atmosphere. Heating the product to about 900°C eliminates the reaction by-products, which include hydrofluoric acid, sulfur trioxide, and phosphoric anhydride. Heating it to higher temperatures in an inert atmosphere decomposes Pa(PO3)4 into the diphosphate PaP2O7, which is analogous to diphosphates of other actinides. In the diphosphate, the PO3 groups form pyramids of C2v symmetry. Heating PaP2O7 in air to 1400°C decomposes it into the pentoxides of phosphorus and protactinium.[61]
Halides
Protactinium(V) fluoride is a white compound that forms tetragonal crystals, isomorphic to β-UF5.[69] Protactinium(V) chloride forms yellow crystals where protactinium ions are arranged in pentagonal bipyramids and coordinated by 7 other ions. The coordination changes to octahedral in the brown protactinium(V) bromide, but is unknown for protactinium(V) iodide. The protactinium coordination in all its tetrahalides is 8, but the arrangement is square antiprismatic in protactinium(IV) fluoride and dodecahedral in the chloride and bromide. Brown-colored protactinium(III) iodide has been reported, where protactinium ions are 8-coordinated in a bicapped trigonal prismatic arrangement.[70]
Coordination of protactinium (solid circles) and chlorine atoms (open circles) in protactinium(V) chloride.
Protactinium(V) chloride has a polymeric structure of monoclinic symmetry.[71] There, within one polymeric chain, all chlorine atoms lie in one graphite-like plane and form planar pentagons around the protactinium ions. The 7-coordination of protactinium originates from the five chlorine atoms and two bonds to protactinium atoms belonging to the nearby chains. It easily hydrolyzes in water.[72] It melts at 300°C and sublimates at even lower temperatures.
Protactinium(V) fluoride can be prepared by reacting protactinium oxide with either bromine pentafluoride or bromine trifluoride at about 600°C, and protactinium(IV) fluoride is obtained from the oxide and a mixture of hydrogen and hydrogen fluoride at 600°C; a large excess of hydrogen is required to remove atmospheric oxygen leaks into the reaction.[46]
Protactinium(V) chloride is prepared by reacting protactinium oxide with carbon tetrachloride at temperatures of 200–300°C.[46] The by-products (such as PaOCl3) are removed by fractional sublimation.[59] Reduction of protactinium(V) chloride with hydrogen at about 800°C yields protactinium(IV) chloride – a yellow-green solid that sublimes in vacuum at 400°C. It can also be obtained directly from protactinium dioxide by treating it with carbon tetrachloride at 400°C.[46]
Protactinium bromides are produced by the action of aluminium bromide, hydrogen bromide, carbon tetrabromide, or a mixture of hydrogen bromide and thionyl bromide on protactinium oxide. They can alternatively be produced by reacting protactinium pentachloride with hydrogen bromide or thionyl bromide.[46] Protactinium(V) bromide has two similar monoclinic forms: one is obtained by sublimation at 400–410°C, and another by sublimation at a slightly lower temperature of 390–400°C.[58][60]
Protactinium iodides can be produced by reacting protactinium metal with elemental iodine at 600°C, and by reacting Pa2O5 with AlI3 at elevated temperatures.[46]Protactinium(III) iodide can be obtained by heating protactinium(V) iodide in vacuum.[72] As with oxides, protactinium forms mixed halides with alkali metals. The most remarkable among these is Na3PaF8, where the protactinium ion is symmetrically surrounded by 8 F− ions, forming a nearly perfect cube.[73]
More complex protactinium fluorides are also known, such as Pa2F9[72] and ternary fluorides of the types MPaF6 (M = Li, Na, K, Rb, Cs or NH4), M2PaF7 (M = K, Rb, Cs or NH4), and M3PaF8 (M = Li, Na, Rb, Cs), all of which are white crystalline solids. The MPaF6 formula can be represented as a combination of MF and PaF5. These compounds can be obtained by evaporating a hydrofluoric acid solution containing both complexes. For the small alkali cations like Na, the crystal structure is tetragonal, whereas it becomes orthorhombic for larger cations K+, Rb+, Cs+ or NH4+. A similar variation was observed for the M2PaF7 fluorides: namely, the crystal symmetry was dependent on the cation and differed for Cs2PaF7 and M2PaF7 (M = K, Rb or NH4).[53]
Other inorganic compounds
Oxyhalides and oxysulfides of protactinium are known. PaOBr3 has a monoclinic structure composed of double-chain units where protactinium has coordination 7 and is arranged into pentagonal bipyramids. The chains are interconnected through oxygen and bromine atoms, and each oxygen atom is related to three protactinium atoms.[58] PaOS is a light-yellow, non-volatile solid with a cubic crystal lattice isostructural to that of other actinide oxysulfides. It is obtained by reacting protactinium(V) chloride with a mixture of hydrogen sulfide and carbon disulfide at 900°C.[46]
In hydrides and nitrides, protactinium has a low oxidation state of about +3. The hydride is obtained by direct action of hydrogen on the metal at 250°C, and the nitride is a product of ammonia and protactinium tetrachloride or pentachloride. This bright yellow solid is thermally stable to 800°C in vacuum. Protactinium carbide (PaC) is formed by the reduction of protactinium tetrafluoride with barium in a carbon crucible at a temperature of about 1400°C.[46] Protactinium forms borohydrides, which include Pa(BH4)4. It has an unusual polymeric structure with helical chains, where the protactinium atom has coordination number of 14 and is surrounded by six BH4− ions.[74]
Organometallic compounds
The proposed structure of the protactinocene (Pa(C8H8)2) molecule
Protactinium(IV) forms a tetrahedral complex tetrakis(cyclopentadienyl)protactinium(IV) (or Pa(C5H5)4) with four cyclopentadienyl rings, which can be synthesized by reacting protactinium(IV) chloride with Be(C5H5)2. One ring can be substituted with a halide atom.[75] Another organometallic complex is the golden-yellow bis(π-cyclooctatetraene) protactinium, or protactinocene (Pa(C8H8)2), which is analogous in structure to uranocene. There, the metal atom is sandwiched between two cyclooctatetraene ligands. Similar to uranocene, it can be prepared by reacting protactinium tetrachloride with dipotassium cyclooctatetraenide (K2C8H8) in tetrahydrofuran.[62]
Applications
Although protactinium is situated in the periodic table between uranium and thorium, both of which have numerous applications, there are currently no uses for protactinium outside scientific research owing to its scarcity, high radioactivity, and high toxicity.[38]
231Pa arises naturally from the decay of natural 235U, and artificially in nuclear reactors by the reaction 232Th+n→231Th+2n and the subsequent beta decay of 231Th. It was once thought to be able to support a nuclear chain reaction, which could in principle be used to build nuclear weapons; the physicistWalter Seifritz[de] once estimated the associated critical mass as 750±180kg.[76] However, the possibility of criticality of 231Pa has since been ruled out.[36][77]
With the advent of highly sensitive mass spectrometers, an application of 231Pa as a tracer in geology and paleoceanography has become possible. In this application, the ratio of 231Pa to 230Th is used for radiometric dating of sediments which are up to 175,000 years old, and in modeling of the formation of minerals.[39] In particular, its evaluation in oceanic sediments helped to reconstruct the movements of North Atlantic water bodies during the last melting of Ice Ageglaciers.[78] Some of the protactinium-related dating variations rely on analysis of the relative concentrations of several long-living members of the uranium decay chain – uranium, protactinium, and thorium, for example. These elements have 6, 5, and 4 valence electrons, thus favoring +6, +5, and +4 oxidation states respectively, and display different physical and chemical properties. Thorium and protactinium, but not uranium compounds, are poorly soluble in aqueous solutions and precipitate into sediments; the precipitation rate is faster for thorium than for protactinium. The concentration analysis for both protactinium-231 (half-life 32,760 years) and 230Th (half-life 75,380 years) improves measurement accuracy compared to when only one isotope is measured; this double-isotope method is also weakly sensitive to inhomogeneities in the spatial distribution of the isotopes and to variations in their precipitation rate.[39][79]
Precautions
Protactinium is both toxic and highly radioactive; thus, it is handled exclusively in a sealed glove box. Its major isotope 231Pa has a specific activity of 0.048 curies (1.8GBq) per gram and primarily emits alpha particles, which can be stopped by a thin layer of any material. However, it slowly decays into 227Ac, and then follows the more rapid actinium series, making its total activity (alpha, beta, and gamma) greater than one would calculate from that figure.
As protactinium is present in small amounts in most natural products and materials, it is ingested with food or water and inhaled with air. Only about 0.05% of ingested protactinium is absorbed into the blood and the remainder is excreted. From the blood, about 40% of the protactinium deposits in the bones, about 15% goes to the liver, 2% to the kidneys, and the rest leaves the body. The biological half-life of protactinium is about 50 years in the bones, whereas its biological half-life in other organs has a fast and slow component. For example, 70% of the protactinium in the liver has a biological half-life of 10 days, and the remaining 30% for 60 days. The corresponding values for kidneys are 20% (10 days) and 80% (60 days). In each affected organ, protactinium promotes cancer via its radioactivity.[38][68] The maximum amount of Pa allowed in the human body is 0.03μCi (1.1kBq), which corresponds to 0.5 micrograms of 231Pa.[80] The maximum allowed concentrations of 231Pa in the air in Germany is 3×10−4Bq/m3.[68]
Ada Hitchins, who helped Soddy in discovering the element protactinium
Notes
↑The prefix "eka" is derived from the Sanskrit एक, meaning "one" or "first." In chemistry, it was formerly used to denote an element one period below the element name following it.
123Arblaster, John W. (2018). Selected Values of the Crystallographic Properties of Elements. Materials Park, Ohio: ASM International. ISBN978-1-62708-155-9.
12Meitner, Lise (1918). "Die Muttersubstanz des Actiniums, Ein Neues Radioaktives Element von Langer Lebensdauer". Zeitschrift für Elektrochemie und angewandte physikalische Chemie. 24 (11–12): 169–173. doi:10.1002/bbpc.19180241107. ISSN0372-8323.
↑"Protactinium"(PDF). Human Health Fact Sheet. ANL (Argonne National Laboratory). November 2001. Retrieved 4 September 2023. The name comes from the Greek work protos (meaning first) and the element actinium, because protactinium is the precursor of actinium.
↑National Research Council (U.S.). Conference on Glossary of Terms in Nuclear Science and Technology (1957). A Glossary of Terms in Nuclear Science and Technology. American Society of Mechanical Engineers. p.180. Retrieved 25 July 2015.
12Zhang, M. M.; Wang, J. G.; Ma, L.; Gan, Z. G.; Zhang, Z. Y.; Huang, M. H.; Yang, H. B.; Yang, C. L.; Andreyev, A. N.; Yuan, C. X.; Tian, Y. L.; Wang, Y. S.; Wang, J. Y.; Qiang, Y. H.; Wu, X. L.; Xu, S. Y.; Zhao, Z.; Huang, X. Y.; Li, Z. C.; Zhou, H.; Zhang, X.; Xie, G.; Zhu, L.; Guan, F.; Zheng, J. H.; Sun, L. C.; Li, Y. J.; Yang, H. R.; Duan, L. M.; Lu, Z. W.; Huang, W. X.; Sun, L. T.; He, Y.; Xu, H. S.; Niu, Y. F.; He, X. T.; Ren, Z. Z.; Zhou, S. G. (29 May 2025). "Discovery of the α-emitting isotope 210Pa". Nature Communications. 16 (1): 5003. doi:10.1038/s41467-025-60047-2. ISSN2041-1723. PMC12123024. PMID40442068.
12Asprey, L. B.; Kruse, F. H.; Rosenzweig, A.; Penneman, R. A. (1966). "Synthesis and X-Ray Properties of Alkali Fluoride-Protactinium Pentafluoride Complexes". Inorganic Chemistry. 5 (4): 659. doi:10.1021/ic50038a034.
↑Di Giandomenico, M.V.; Le Naour, C. (2009). "Complex formation between protactinium(V) and sulfate ions at 10 and 60°C". Inorganica Chimica Acta. 362 (9): 3253–3258. doi:10.1016/j.ica.2009.02.033.
↑Eitrheim, Eric S.; Knight, Andrew W.; Schultz, Michael K.; Forbes, Tori Z.; Nelson, Andrew W. (2017). "Recent Advancements in the Radiochemistry of Elements Pertaining to Select Nuclear Materials and Wastes". ACS Symposium Series. Vol.1263. Washington, DC: American Chemical Society. doi:10.1021/bk-2017-1263.ch009. ISBN978-0-8412-3255-6.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.