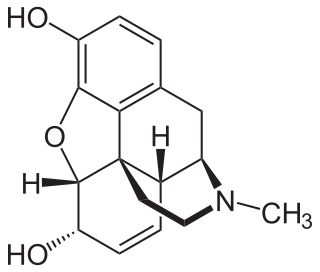
Morphine, formerly also called morphia, is an opiate that is found naturally in opium, a dark brown resin produced by drying the latex of opium poppies. It is mainly used as an analgesic. There are numerous methods used to administer morphine: orally; administered under the tongue; via inhalation; injection into a vein, injection into a muscle, injection under the skin, or injection into the spinal cord area; transdermal; or via administered into the rectal canal suppository. It acts directly on the central nervous system (CNS) to induce analgesia and alter perception and emotional response to pain. Physical and psychological dependence and tolerance may develop with repeated administration. It can be taken for both acute pain and chronic pain and is frequently used for pain from myocardial infarction, kidney stones, and during labor. Its maximum effect is reached after about 20 minutes when administered intravenously and 60 minutes when administered by mouth, while the duration of its effect is 3–7 hours. Long-acting formulations of morphine are sold under the brand names MS Contin and Kadian, among others. Generic long-acting formulations are also available.

Dihydrocodeine is a semi-synthetic opioid analgesic prescribed for pain or severe dyspnea, or as an antitussive, either alone or compounded with paracetamol (acetaminophen) or aspirin. It was developed in Germany in 1908 and first marketed in 1911.
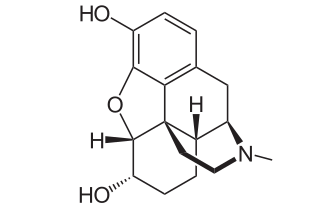
Dihydromorphine is a semi-synthetic opioid structurally related to and derived from morphine. The 7,8-double bond in morphine is reduced to a single bond to get dihydromorphine. Dihydromorphine is a moderately strong analgesic and is used clinically in the treatment of pain and also is an active metabolite of the analgesic opioid drug dihydrocodeine. Dihydromorphine occurs in trace quantities in assays of opium on occasion, as does dihydrocodeine, dihydrothebaine, tetrahydrothebaine, etc. The process for manufacturing dihydromorphine from morphine for pharmaceutical use was developed in Germany in the late 19th century, with the synthesis being published in 1900 and the drug introduced clinically as Paramorfan shortly thereafter. A high-yield synthesis from tetrahydrothebaine was later developed.

6-Monoacetylmorphine is an opioid and also one of three active metabolites of heroin (diacetylmorphine), the others being morphine and the much less active 3-monoacetylmorphine (3-MAM).

Diacetyldihydromorphine is a potent opiate derivative developed in Germany in 1928 which is rarely used in some countries for the treatment of severe pain such as that caused by terminal cancer, as another form of diacetylmorphine. Diacetyldihydromorphine is fast-acting and longer-lasting than diamorphine, with a duration of action of around 4–7 hours.

The μ-opioid receptors (MOR) are a class of opioid receptors with a high affinity for enkephalins and beta-endorphin, but a low affinity for dynorphins. They are also referred to as μ(mu)-opioid peptide (MOP) receptors. The prototypical μ-opioid receptor agonist is morphine, the primary psychoactive alkaloid in opium and for which the receptor was named, with mu being the first letter of Morpheus, the compound's namesake in the original Greek. It is an inhibitory G-protein coupled receptor that activates the Gi alpha subunit, inhibiting adenylate cyclase activity, lowering cAMP levels.

Ohmefentanyl is an extremely potent opioid analgesic drug which selectively binds to the μ-opioid receptor.

Lofentanil or lofentanyl is one of the most potent opioid analgesics known and is an analogue of fentanyl, which was developed in 1960. It is most similar to the highly potent opioid carfentanil (4-carbomethoxyfentanyl), only slightly more potent. Lofentanil can be described as 3-methylcarfentanil, or 3-methyl-4-carbomethoxyfentanyl. While 3-methylfentanyl is considerably more potent than fentanyl itself, lofentanil is only slightly stronger than carfentanil. This suggests that substitution at both the 3 and 4 positions of the piperidine ring introduces steric hindrance which prevents μ-opioid affinity from increasing much further. As with other 3-substituted fentanyl derivatives such as ohmefentanyl, the stereoisomerism of lofentanil is very important, with some stereoisomers being much more potent than others.
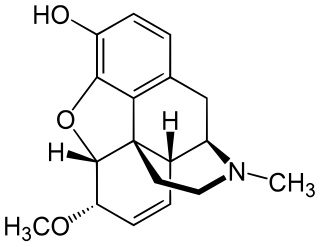
Heterocodeine (6-methoxymorphine) is an opiate derivative, the 6-methyl ether of morphine, and a structural isomer of codeine; it is called "hetero-" because it is the reverse isomer of codeine. Heterocodeine was first synthesised in 1932 and first patented in 1935. It can be made from morphine by selective methylation. Codeine is the natural mono-methyl ether, but must be metabolized for activity. In contrast the semi-synthetic mono-methyl ether, heterocodeine is a direct agonist. The 6,7,8,14 tetradehydro 3,6 methyl di-ether of morphine is thebaine.

RB-101 is a drug that acts as an enkephalinase inhibitor, which is used in scientific research.
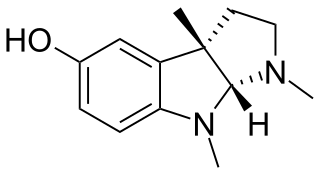
Eseroline is a drug which acts as an opioid agonist. It is a metabolite of the acetylcholinesterase inhibitor physostigmine but unlike physostigmine, the acetylcholinesterase inhibition produced by eseroline is weak and easily reversible, and it produces fairly potent analgesic effects mediated through the μ-opioid receptor. This mixture of activities gives eseroline an unusual pharmacological profile, although its uses are limited by side effects such as respiratory depression and neurotoxicity.

Azaprocin is a drug which is an opioid analgesic with approximately ten times the potency of morphine, and a fast onset and short duration of action. It was discovered in 1963, but has never been marketed.

SC-17599 is a steroid derivative drug discovered in 1968 which acts as a selective μ-opioid receptor agonist, with little or no affinity for the δ-opioid or κ-opioid receptors. It is an active analgesic in vivo, more potent than codeine or pethidine but slightly less potent than morphine, and produces similar effects to morphine in animals but with less sedation
An equianalgesic chart is a conversion chart that lists equivalent doses of analgesics. Equianalgesic charts are used for calculation of an equivalent dose between different analgesics. Tables of this general type are also available for NSAIDs, benzodiazepines, depressants, stimulants, anticholinergics and others.
JDTic is a selective, long-acting ("inactivating") antagonist of the κ-opioid receptor (KOR). JDTic is a 4-phenylpiperidine derivative, distantly related structurally to analgesics such as pethidine and ketobemidone, and more closely to the MOR antagonist alvimopan. In addition, it is structurally distinct from other KOR antagonists such as norbinaltorphimine. JDTic has been used to create crystal structures of KOR [ PDB: 4DJH, 6VI4].

Pregnenolone sulfate is an endogenous excitatory neurosteroid that is synthesized from pregnenolone. It is known to have cognitive and memory-enhancing, antidepressant, anxiogenic, and proconvulsant effects.

1-Iodomorphine is a semi-synthetic narcotic analgesic formed by halogenation of the 1 position on the morphine carbon skeleton. Halogenated morphine derivatives were first synthesised in Germany, Austria/Austria-Hungary, the United Kingdom and the United States in the period 1890 to 1930. Use of this drug increased after 1945 for the below-mentioned research. It is a research chemical which is often prepared in the laboratory when it is needed.

SB-206553 is a drug which acts as a mixed antagonist for the 5-HT2B and 5-HT2C serotonin receptors. It has anxiolytic properties in animal studies and interacts with a range of other drugs. It has also been shown to act as a positive allosteric modulator of α7 nicotinic acetylcholine receptors. Modified derivatives of SB-206553 have been used to probe the structure of the 5-HT2B receptor.

Biphalin is a dimeric enkephalin endogenous peptide (Tyr-D-Ala-Gly-Phe-NH)2 composed of two tetrapeptides derived from enkephalins, connected 'tail-to-tail' by a hydrazide bridge. The presence of two distinct pharmacophores confers on biphalin a high affinity for both μ and δ opioid receptors (with an EC50 of about 1–5 nM for both μ and δ receptors), therefore it has analgesic activity. Biphalin presents a considerable antinociceptive profile. In fact, when administered intracerebroventricularly in mice, biphalin displays a potency almost 7-fold greater than that of the ultra-potent alkaloid agonist, etorphine and 7000-fold greater than morphine; biphalin and morphine were found to be equipotent after intraperitoneal administration. The extraordinary in vivo potency shown by this compound is coupled with low side-effects, in particular, to produce no dependency in chronic use. For these reasons, several efforts have been carried out in order to obtain more information about structure-activity relationship (SAR). Results clearly indicate that, at least for μ receptor binding, the presence of two pharmacophores is not necessary; Tyr1 is indispensable for analgesic activity, while replacing Phe at the position 4 and 4' with non-aromatic, but lipophilic amino acids does not greatly change the binding properties and in general 4,4' positions are found to be important to design biphalin analogues with increased potency and modified μ/δ selectivity. The hydrazide linker is not fundamental for activity or binding, and it can be conveniently substituted by different conformationally constrained cycloaliphatic diamine linkers.

Cannabidiol-dimethylheptyl (CBD-DMH or DMH-CBD) is a synthetic homologue of cannabidiol where the pentyl chain has been replaced by a dimethylheptyl chain. Several isomers of this compound are known. The most commonly used isomer in research is (−)-CBD-DMH, which has the same stereochemistry as natural cannabidiol, and a 1,1-dimethylheptyl side chain. This compound is not psychoactive and acts primarily as an anandamide reuptake inhibitor, but is more potent than cannabidiol as an anticonvulsant and has around the same potency as an antiinflammatory. Unexpectedly the “unnatural” enantiomer (+)-CBD-DMH, which has reversed stereochemistry from cannabidiol, was found to be a directly acting cannabinoid receptor agonist with a Ki of 17.4nM at CB1 and 211nM at CB2, and produces typical cannabinoid effects in animal studies, as does its 7-OH derivative.



