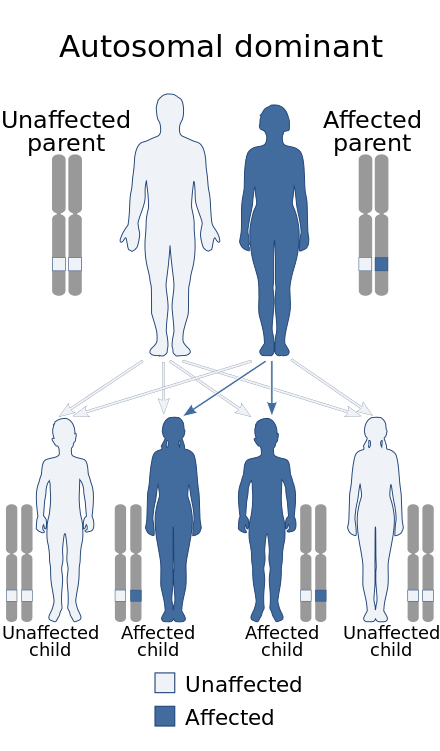
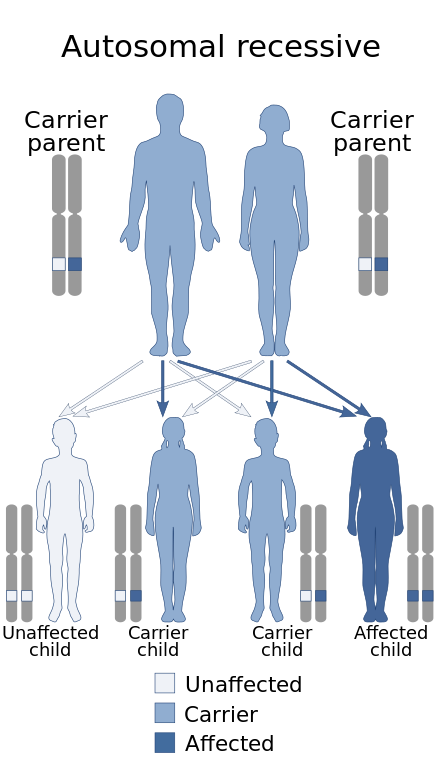
A genetic disorder is a health problem caused by one or more abnormalities in the genome. It can be caused by a mutation in a single gene (monogenic) or multiple genes (polygenic) or by a chromosomal abnormality. Although polygenic disorders are the most common, the term is mostly used when discussing disorders with a single genetic cause, either in a gene or chromosome. The mutation responsible can occur spontaneously before embryonic development, or it can be inherited from two parents who are carriers of a faulty gene or from a parent with the disorder. Some disorders are caused by a mutation on the X chromosome and have X-linked inheritance. Very few disorders are inherited on the Y chromosome or mitochondrial DNA.

Muscular dystrophy (MD) is a group of muscle diseases that results in increasing weakening and breakdown of skeletal muscles over time. The disorders differ in which muscles are primarily affected, the degree of weakness, how fast they worsen, and when symptoms begin. Many people will eventually become unable to walk. Some types are also associated with problems in other organs.

Arthrogryposis multiplex congenita (AMC), or simply arthrogryposis, describes congenital joint contracture in two or more areas of the body. It derives its name from Greek, literally meaning "curving of joints".

Becker muscular dystrophy is an X-linked recessive inherited disorder characterized by slowly progressing muscle weakness of the legs and pelvis. It is a type of dystrophinopathy. This is caused by mutations in the dystrophin gene, which encodes the protein dystrophin. Becker muscular dystrophy is related to Duchenne muscular dystrophy in that both result from a mutation in the dystrophin gene, but has a milder course.

Oculopharyngeal muscular dystrophy (OPMD) is a rare form of muscular dystrophy with symptoms generally starting when an individual is 40 to 50 years old. It can be autosomal dominant neuromuscular disease or autosomal recessive. The most common inheritance of OPMD is autosomal dominant, which means only one copy of the mutated gene needs to be present in each cell. Children of an affected parent have a 50% chance of inheriting the mutant gene.

Fukuyama congenital muscular dystrophy (FCMD) is a rare, autosomal recessive form of muscular dystrophy mainly described in Japan but also identified in Turkish and Ashkenazi Jewish patients; fifteen cases were first described on 1960 by Dr. Yukio Fukuyama.
Nemaline myopathy is a congenital, hereditary neuromuscular disorder with many symptoms that can occur such as muscle weakness, hypoventilation, swallowing dysfunction, and impaired speech ability. The severity of these symptoms varies and can change throughout one's life to some extent. The prevalence is estimated at 1 in 50,000 live births. It is the most common non-dystrophic myopathy.
Hereditary inclusion body myopathies (HIBM) are a group of rare genetic disorders which have different symptoms. Generally, they are neuromuscular disorders characterized by muscle weakness developing in young adults. Hereditary inclusion body myopathies comprise both autosomal recessive and autosomal dominant muscle disorders that have a variable expression (phenotype) in individuals, but all share similar structural features in the muscles.
Congenital muscular dystrophies are autosomal recessively-inherited muscle diseases. They are a group of heterogeneous disorders characterized by muscle weakness which is present at birth and the different changes on muscle biopsy that ranges from myopathic to overtly dystrophic due to the age at which the biopsy takes place.
The sarcoglycanopathies are a collection of diseases resulting from mutations in any of the five sarcoglycan genes: α, β, γ, δ or ε. The five sarcoglycanopathies are: α-sarcoglycanopathy, LGMD2D; β-sarcoglycanopathy, LGMD2E; γ-sarcoglycanopathy, LGMD2C; δ-sarcoglycanopathy, LGMD2F and ε-sarcoglycanopathy, myoclonic dystonia. The four different sarcoglycan genes encode proteins that form a tetrameric complex at the muscle cell plasma membrane. This complex stabilizes the association of dystrophin with the dystroglycans and contributes to the stability of the plasma membrane cytoskeleton. The four sarcoglycan genes are related to each other structurally and functionally, but each has a distinct chromosome location.
The sarcoglycans are a family of transmembrane proteins involved in the protein complex responsible for connecting the muscle fibre cytoskeleton to the extracellular matrix, preventing damage to the muscle fibre sarcolemma through shearing forces.

Emery–Dreifuss muscular dystrophy is a condition that mainly affects muscles used for movement, such as skeletal muscles and also affects the cardiac muscle, it is named after Alan Eglin H. Emery and Fritz E. Dreifuss.

Beta-sarcoglycan is a protein that in humans is encoded by the SGCB gene.

Delta-sarcoglycan is a protein that in humans is encoded by the SGCD gene.

Alpha-sarcoglycan is a protein that in humans is encoded by the SGCA gene.

Gamma-sarcoglycan is a protein that in humans is encoded by the SGCG gene. The α to δ-sarcoglycans are expressed predominantly (β) or exclusively in striated muscle. A mutation in any of the sarcoglycan genes may lead to a secondary deficiency of the other sarcoglycan proteins, presumably due to destabilisation of the sarcoglycan complex. The disease-causing mutations in the α to δ genes cause disruptions within the dystrophin-associated protein (DAP) complex in the muscle cell membrane. The transmembrane components of the DAP complex link the cytoskeleton to the extracellular matrix in adult muscle fibres, and are essential for the preservation of the integrity of the muscle cell membrane.

Myoclonic dystonia or Myoclonus dystonia syndrome is a rare movement disorder that induces spontaneous muscle contraction causing abnormal posture. The prevalence of myoclonus dystonia has not been reported, however, this disorder falls under the umbrella of movement disorders which affect thousands worldwide. Myoclonus dystonia results from mutations in the SGCE gene coding for an integral membrane protein found in both neurons and muscle fibers. Those suffering from this disease exhibit symptoms of rapid, jerky movements of the upper limbs (myoclonus), as well as distortion of the body's orientation due to simultaneous activation of agonist and antagonist muscles (dystonia).
Ullrich congenital muscular dystrophy is a form of congenital muscular dystrophy. It is associated with variants of type VI collagen, it is commonly associated with muscle weakness and respiratory problems, though cardiac issues are not associated with this type of CMD. It is named after Otto Ullrich, who is also known for the Ullrich-Turner syndrome.
Anoctamin 5 (ANO5) is a protein that in humans is encoded by the ANO5 gene.
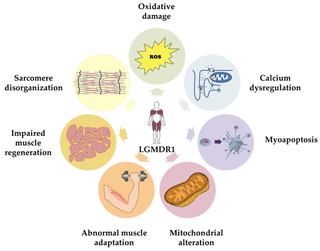
Calpainopathy is the most common type of autosomal recessive limb-girdle muscular dystrophy (LGMD). It has a predisposition for affecting the muscles of the hip girdle and shoulder girdle.