Dynorphins (Dyn) are a class of opioid peptides that arise from the precursor protein prodynorphin. When prodynorphin is cleaved during processing by proprotein convertase 2 (PC2), multiple active peptides are released: dynorphin A, dynorphin B, and α/β-neoendorphin. Depolarization of a neuron containing prodynorphin stimulates PC2 processing, which occurs within synaptic vesicles in the presynaptic terminal. Occasionally, prodynorphin is not fully processed, leading to the release of "big dynorphin". "Big dynorphin" is a 32-amino acid molecule consisting of both dynorphin A and dynorphin B.

An opioid antagonist, or opioid receptor antagonist, is a receptor antagonist that acts on one or more of the opioid receptors.
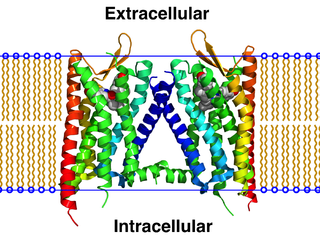
The κ-opioid receptor or kappa opioid receptor, abbreviated KOR or KOP for its ligand ketazocine, is a G protein-coupled receptor that in humans is encoded by the OPRK1 gene. The KOR is coupled to the G protein Gi/G0 and is one of four related receptors that bind opioid-like compounds in the brain and are responsible for mediating the effects of these compounds. These effects include altering nociception, consciousness, motor control, and mood. Dysregulation of this receptor system has been implicated in alcohol and drug addiction.

The μ-opioid receptors (MOR) are a class of opioid receptors with a high affinity for enkephalins and beta-endorphin, but a low affinity for dynorphins. They are also referred to as μ(mu)-opioid peptide (MOP) receptors. The prototypical μ-opioid receptor agonist is morphine, the primary psychoactive alkaloid in opium and for which the receptor was named, with mu being the first letter of Morpheus, the compound's namesake in the original Greek. It is an inhibitory G-protein coupled receptor that activates the Gi alpha subunit, inhibiting adenylate cyclase activity, lowering cAMP levels.

Nalfurafine is an antipruritic that is marketed in Japan for the treatment of uremic pruritus in individuals with chronic kidney disease undergoing hemodialysis. It activates the κ-opioid receptor (KOR) and is potent, selective, and centrally active. It was the first selective KOR agonist approved for clinical use. It has also been dubiously referred to as the "first non-narcotic opioid drug" in history.
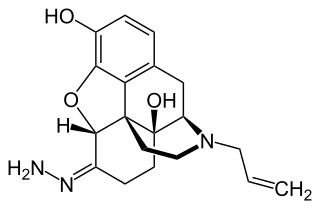
Naloxazone is an irreversible μ-opioid receptor antagonist which is selective for the μ1 receptor subtype. Naloxazone produces very long lasting antagonist effects as it forms a covalent bond to the active site of the μ-opioid receptor, thus making it impossible for the molecule to unbind and blocking the receptor permanently until the receptor is recycled by endocytosis.

Oxymorphazone is an opioid analgesic drug related to oxymorphone. Oxymorphazone is a potent and long acting μ-opioid agonist which binds irreversibly to the receptor, forming a covalent bond which prevents it from detaching once bound. This gives it an unusual pharmacological profile, and while oxymorphazone is only around half the potency of oxymorphone, with higher doses the analgesic effect becomes extremely long lasting, with a duration of up to 48 hours. However, tolerance to analgesia develops rapidly with repeated doses, as chronically activated opioid receptors are rapidly internalised by β-arrestins, similar to the results of non-covalent binding by repeated doses of agonists with extremely high binding affinity such as lofentanil.

β-Chlornaltrexamine (β-CNA) is a non-selective irreversible antagonist of the μ-opioid receptor (MOR), the δ-opioid receptor (DOR), and the κ-opioid receptor (KOR), which forms a covalent bond to the binding sites of these receptors and has ultra-long-lasting opioid antagonist effects. Although it is predominantly antagonistic, β-CNA also shows some irreversible mixed agonist–antagonist activity at the MOR and KOR and some associated analgesic effects. Its alkylating group is a bis(chloroalkyl)amino-residue similar to that of the nitrogen mustards.
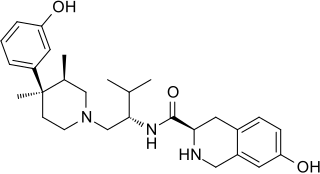
JDTic is a selective, long-acting ("inactivating") antagonist of the κ-opioid receptor (KOR). JDTic is a 4-phenylpiperidine derivative, distantly related structurally to analgesics such as pethidine and ketobemidone, and more closely to the MOR antagonist alvimopan. In addition, it is structurally distinct from other KOR antagonists such as norbinaltorphimine. JDTic has been used to create crystal structures of KOR [ PDB: 4DJH, 6VI4].

Alazocine, also known more commonly as N-allylnormetazocine (NANM), is a synthetic opioid analgesic of the benzomorphan family related to metazocine, which was never marketed. In addition to its opioid activity, the drug is a sigma receptor agonist, and has been used widely in scientific research in studies of this receptor. Alazocine is described as a potent analgesic, psychotomimetic or hallucinogen, and opioid antagonist. Moreover, one of its enantiomers was the first compound that was found to selectively label the σ1 receptor, and led to the discovery and characterization of the receptor.

IBNtxA, or 3-iodobenzoyl naltrexamine, is an atypical opioid analgesic drug derived from naltrexone. In animal studies it produces potent analgesic effects that are blocked by levallorphan and so appear to be μ-opioid mediated, but it fails to produce constipation or respiratory depression, and is neither rewarding or aversive in conditioned place preference protocols. These unusual properties are thought to result from agonist action at a splice variant or heterodimer of the μ-opioid receptor, rather than at the classical full length form targeted by conventional opioid drugs.

Clocinnamox is a selective and irreversible antagonist of the μ-opioid receptor. Closely related compounds include methocinnamox (MCAM) and methoclocinnamox (MCCAM). They were derived via structural modification of buprenorphine. Clocinnamox was first described in the scientific literature by 1992.

Buprenorphine/samidorphan is a combination formulation of buprenorphine and samidorphan which is under development as an add on to antidepressants in treatment-resistant depression (TRD).
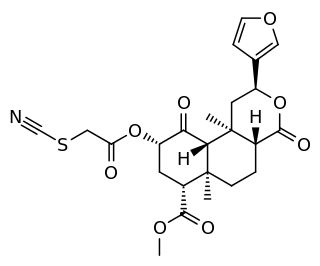
RB-64 is a semi-synthetic derivative of salvinorin A. It is an irreversible agonist, with a reactive thiocyanate group that forms a bond to the κ-opioid receptor (KOR), resulting in very high potency. It is functionally selective, activating G proteins more potently than β-arrestin-2. RB-64 has a bias factor of up to 96 and is analgesic with fewer of the side-effects associated with unbiased KOR agonists. The analgesia is long-lasting. Compared with unbiased agonists, RB-64 evokes considerably less receptor internalization.

Norbuprenorphine-3-glucuronide (N3G) is a major active metabolite of the opioid modulator buprenorphine. It has affinity for the κ-opioid receptor and the nociceptin receptor, but not for the μ- or δ-opioid receptors. Whether N3G acts as an agonist or antagonist of each of the former two respective sites has yet to be determined. In animals, N3G has been found to produce sedation, decreased locomotion, and a small amount of antinociception, properties which are consistent with the effects of κ-opioid receptor agonists. In addition, N3G has been found to reduce tidal volume but not respiratory rate. Unlike norbuprenorphine, but similarly to buprenorphine and buprenorphine-3-glucuronide, N3G is not a substrate for P-glycoprotein. However, due to its highly hydrophilic nature, N3G nonetheless passes the blood-brain-barrier in only very small amounts.

6β-Naltrexol, or 6β-hydroxynaltrexone, is a peripherally-selective opioid receptor antagonist related to naltrexone. It is a major active metabolite of naltrexone formed by hepatic dihydrodiol dehydrogenase enzymes. With naltrexone therapy, 6β-naltrexol is present at approximately 10- to 30-fold higher concentrations than naltrexone at steady state due to extensive first-pass metabolism of naltrexone into 6β-naltrexol. In addition to being an active metabolite of naltrexone, 6β-naltrexol was itself studied for the treatment of opioid-induced constipation. It was found to be effective and well-tolerated, and did not precipitate opioid withdrawal symptoms or interfere with opioid pain relief, but development was not further pursued.
Peripherally selective drugs have their primary mechanism of action outside of the central nervous system (CNS), usually because they are excluded from the CNS by the blood–brain barrier. By being excluded from the CNS, drugs may act on the rest of the body without producing side-effects related to their effects on the brain or spinal cord. For example, most opioids cause sedation when given at a sufficiently high dose, but peripherally selective opioids can act on the rest of the body without entering the brain and are less likely to cause sedation. These peripherally selective opioids can be used as antidiarrheals, for instance loperamide (Imodium).
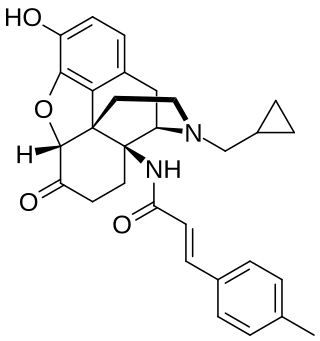
Methocinnamox (MCAM) is an opioid receptor antagonist. It is a pseudo-irreversible non-competitive antagonist of the μ-opioid receptor and a competitive antagonist of the κ- and δ-opioid receptors. The drug has a very long duration of action of up to months with a single dose due to its pseudo-irreversibility. It is administered in animals by intravenous or subcutaneous injection.

Methoclocinnamox is a selective pseudo-irreversible partial agonist of the μ-opioid receptor (MOR). It shows a mixture of opioid agonist- and antagonist-like effects. The drug has long-lasting effects and is insurmountable by other MOR ligands.
Naltrexamine, or β-naltrexamine, is an opioid receptor antagonist related to naltrexol and naltrexone. It has served as a parent pharmacophore for irreversible antagonists of the μ-opioid receptor (MOR) such as β-chlornaltrexamine (β-CNA) and β-funaltrexamine (β-FNA). Naltrexamine itself is a neutral antagonist of the MOR and the δ-opioid receptor (DOR) with similarly high affinity for both receptors.
