
Buprenorphine, sold under the brand name Subutex among others, is an opioid used to treat opioid use disorder, acute pain, and chronic pain. It can be used under the tongue (sublingual), in the cheek (buccal), by injection, as a skin patch (transdermal), or as an implant. For opioid use disorder, the patient must have moderate opioid withdrawal symptoms before buprenorphine can be administered under direct observation of a health-care provider.

Naltrexone, sold under the brand name Revia among others, is a medication primarily used to manage alcohol use or opioid use disorder by reducing cravings and feelings of euphoria associated with substance use disorder. It has also been found effective in the treatment of other addictions and may be used for them off-label. An opioid-dependent person should not receive naltrexone before detoxification. It is taken orally or by injection into a muscle. Effects begin within 30 minutes, though a decreased desire for opioids may take a few weeks to occur.

Norbuprenorphine is a major active metabolite of the opioid modulator buprenorphine. It is a μ-opioid, δ-opioid, and nociceptin receptor full agonist, and a κ-opioid receptor partial agonist. In rats, unlike buprenorphine, norbuprenorphine produces marked respiratory depression but with very little antinociceptive effect. In explanation of these properties, norbuprenorphine has been found to be a high affinity P-glycoprotein substrate, and in accordance, shows very limited blood-brain-barrier penetration.

6-Monoacetylmorphine is an opioid and also one of three active metabolites of heroin (diacetylmorphine), the others being morphine and the much less active 3-monoacetylmorphine (3-MAM).
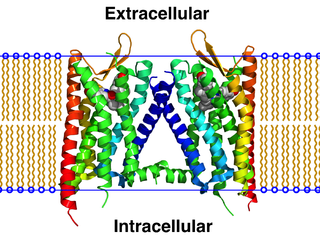
The κ-opioid receptor or kappa opioid receptor, abbreviated KOR or KOP for its ligand ketazocine, is a G protein-coupled receptor that in humans is encoded by the OPRK1 gene. The KOR is coupled to the G protein Gi/G0 and is one of four related receptors that bind opioid-like compounds in the brain and are responsible for mediating the effects of these compounds. These effects include altering nociception, consciousness, motor control, and mood. Dysregulation of this receptor system has been implicated in alcohol and drug addiction.

The μ-opioid receptors (MOR) are a class of opioid receptors with a high affinity for enkephalins and beta-endorphin, but a low affinity for dynorphins. They are also referred to as μ(mu)-opioid peptide (MOP) receptors. The prototypical μ-opioid receptor agonist is morphine, the primary psychoactive alkaloid in opium and for which the receptor was named, with mu being the first letter of Morpheus, the compound's namesake in the original Greek. It is an inhibitory G-protein coupled receptor that activates the Gi alpha subunit, inhibiting adenylate cyclase activity, lowering cAMP levels.

Morphine-3-glucuronide is a metabolite of morphine produced by UGT2B7. It is not active as an opioid agonist, but does have some action as a convulsant, which does not appear to be mediated through opioid receptors, but rather through interaction with glycine and/or GABA receptors. As a polar compound, it has a limited ability to cross the blood–brain barrier, but kidney failure may lead to its accumulation and result in seizures. Probenecid and inhibitors of P-glycoprotein can enhance uptake of morphine-3-glucuronide and, to a lesser extent, morphine-6-glucuronide. Reported side effects related to the accumulation of this metabolite include convulsions, agitation, hallucinations, hyperalgesia, and coma.

Desmetramadol, also known as O-desmethyltramadol (O-DSMT), is an opioid analgesic and the main active metabolite of tramadol. Tramadol is demethylated by the liver enzyme CYP2D6 to desmetramadol in the same way as codeine, and so similarly to the variation in effects seen with codeine, individuals who have a less active form of CYP2D6 will tend to have reduced analgesic effects from tramadol. Because desmetramadol itself does not need to be metabolized to induce an analgesic effect, it can be used in individuals with CYP2D6 inactivating mutations.
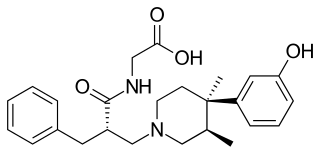
Alvimopan is a drug which behaves as a peripherally acting μ-opioid receptor antagonist. With the limited ability to cross the blood–brain barrier and reach the μ-opioid receptors of the central nervous system, the clinically undesirable effects of centrally acting opioid antagonists are avoided without affecting the intended blockade of μ-opioid receptors in the gastrointestinal tract. It is currently only Food and Drug Administration approved for the treatment of postoperative ileus which it received in May 2008.
An equianalgesic chart is a conversion chart that lists equivalent doses of analgesics. Equianalgesic charts are used for calculation of an equivalent dose between different analgesics. Tables of this general type are also available for NSAIDs, benzodiazepines, depressants, stimulants, anticholinergics and others.
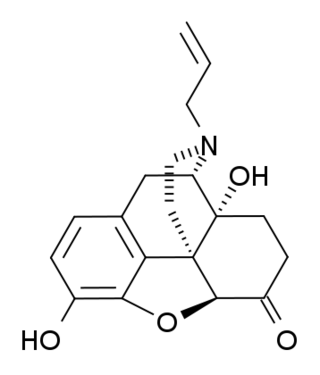
(+)-Naloxone (dextro-naloxone) is a drug which is the opposite enantiomer of the opioid antagonist drug (−)-naloxone. Unlike (−)-naloxone, (+)-naloxone has no significant affinity for opioid receptors, but instead has been discovered to act as a selective antagonist of Toll-like receptor 4. This receptor is involved in immune system responses, and activation of TLR4 induces glial activation and release of inflammatory mediators such as TNF-α and Interleukin-1.

Clocinnamox is a selective and irreversible antagonist of the μ-opioid receptor. Closely related compounds include methocinnamox (MCAM) and methoclocinnamox (MCCAM). They were derived via structural modification of buprenorphine. Clocinnamox was first described in the scientific literature by 1992.

Buprenorphine/samidorphan is a combination formulation of buprenorphine and samidorphan which is under development as an add on to antidepressants in treatment-resistant depression (TRD).
Buprenorphine/naloxone, sold under the brand name Suboxone among others, is a fixed-dose combination medication that includes buprenorphine and naloxone. It is used to treat opioid use disorder, and reduces the mortality of opioid use disorder by 50%. It relieves cravings to use and withdrawal symptoms. Buprenorphine/naloxone is available for use in two different forms, under the tongue or in the cheek.

Norbuprenorphine-3-glucuronide (N3G) is a major active metabolite of the opioid modulator buprenorphine. It has affinity for the κ-opioid receptor and the nociceptin receptor, but not for the μ- or δ-opioid receptors. Whether N3G acts as an agonist or antagonist of each of the former two respective sites has yet to be determined. In animals, N3G has been found to produce sedation, decreased locomotion, and a small amount of antinociception, properties which are consistent with the effects of κ-opioid receptor agonists. In addition, N3G has been found to reduce tidal volume but not respiratory rate. Unlike norbuprenorphine, but similarly to buprenorphine and buprenorphine-3-glucuronide, N3G is not a substrate for P-glycoprotein. However, due to its highly hydrophilic nature, N3G nonetheless passes the blood-brain-barrier in only very small amounts.

Thienorphine is a very potent, extremely long-acting, orally-active opioid analgesic with mixed agonist–antagonist properties which was developed by the Beijing Institute of Pharmacology and Toxicology as a potential treatment for opioid dependence. It is a high-affinity, balanced ligand of the μ-, δ-, and κ-opioid receptors, behaving as a partial agonist of the μ- and κ-opioid receptors and as an antagonist of the δ-opioid receptor. It also possesses relatively low affinity for the nociceptin receptor, where it acts as an antagonist.

Norhydrocodone is the major metabolite of the opioid analgesic hydrocodone. It is formed from hydrocodone in the liver via N-demethylation predominantly by CYP3A4. Unlike hydromorphone, a minor metabolite of hydrocodone, norhydrocodone is described as inactive. However, norhydrocodone is actually an agonist of the μ-opioid receptor with similar potency to hydrocodone, but has been found to produce only minimal analgesia when administered peripherally to animals. This is likely due to poor blood-brain-barrier and thus central nervous system penetration.

Noroxycodone is the major metabolite of the opioid analgesic oxycodone. It is formed from oxycodone in the liver via N-demethylation predominantly by CYP3A4. Noroxycodone binds to and activates the μ-opioid receptor (MOR) similarly to oxycodone, although with one-third of the affinity of oxycodone and 5- to 10-fold lower activational potency. However, although a potent MOR agonist, noroxycodone poorly crosses the blood-brain-barrier into the central nervous system, and for this reason, is only minimally analgesic in comparison.
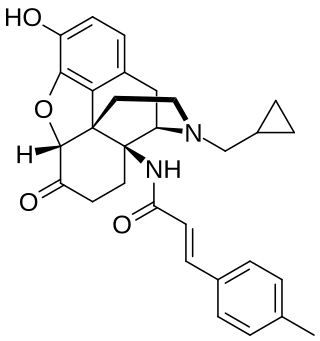
Methocinnamox (MCAM) is an opioid receptor antagonist. It is a pseudo-irreversible non-competitive antagonist of the μ-opioid receptor and a competitive antagonist of the κ- and δ-opioid receptors. The drug has a very long duration of action of up to months with a single dose due to its pseudo-irreversibility. It is administered in animals by intravenous or subcutaneous injection.
