
Leucine (symbol Leu or L) is an essential amino acid that is used in the biosynthesis of proteins. Leucine is an α-amino acid, meaning it contains an α-amino group (which is in the protonated −NH3+ form under biological conditions), an α-carboxylic acid group (which is in the deprotonated −COO− form under biological conditions), and a side chain isobutyl group, making it a non-polar aliphatic amino acid. It is essential in humans, meaning the body cannot synthesize it: it must be obtained from the diet. Human dietary sources are foods that contain protein, such as meats, dairy products, soy products, and beans and other legumes. It is encoded by the codons UUA, UUG, CUU, CUC, CUA, and CUG.
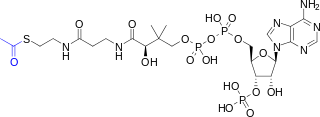
Acetyl-CoA is a molecule that participates in many biochemical reactions in protein, carbohydrate and lipid metabolism. Its main function is to deliver the acetyl group to the citric acid cycle to be oxidized for energy production. Coenzyme A consists of a β-mercaptoethylamine group linked to the vitamin pantothenic acid (B5) through an amide linkage and 3'-phosphorylated ADP. The acetyl group of acetyl-CoA is linked to the sulfhydryl substituent of the β-mercaptoethylamine group. This thioester linkage is a "high energy" bond, which is particularly reactive. Hydrolysis of the thioester bond is exergonic (−31.5 kJ/mol).

Ketogenesis is the biochemical process through which organisms produce ketone bodies through breakdown of fatty acids and ketogenic amino acids. This process supplies energy under circumstances such as fasting or caloric restriction to certain organs, particularly the brain, heart and skeletal muscle. Insufficient gluconeogenesis can cause hypoglycemia and excessive production of ketone bodies, ultimately leading to a life-threatening condition known as non-diabetic ketoacidosis.

3-Hydroxy-3-methylglutaryl-CoA lyase deficiency is an uncommon inherited disorder in which the body cannot properly process the amino acid leucine. Additionally, the disorder prevents the body from making ketones, which are used for energy during fasting.
Numerous genetic disorders are caused by errors in fatty acid metabolism. These disorders may be described as fatty oxidation disorders or as a lipid storage disorders, and are any one of several inborn errors of metabolism that result from enzyme defects affecting the ability of the body to oxidize fatty acids in order to produce energy within muscles, liver, and other cell types.

β-Hydroxybutyric acid, also known as 3-hydroxybutyric acid, is an organic compound and a beta hydroxy acid with the chemical formula CH3CH(OH)CH2CO2H; its conjugate base is β-hydroxybutyrate, also known as 3-hydroxybutyrate. β-Hydroxybutyric acid is a chiral compound with two enantiomers: D-β-hydroxybutyric acid and L-β-hydroxybutyric acid. Its oxidized and polymeric derivatives occur widely in nature. In humans, D-β-hydroxybutyric acid is one of two primary endogenous agonists of hydroxycarboxylic acid receptor 2 (HCA2), a Gi/o-coupled G protein-coupled receptor (GPCR).
β-Hydroxy β-methylglutaryl-CoA (HMG-CoA), also known as 3-hydroxy-3-methylglutaryl-CoA, is an intermediate in the mevalonate and ketogenesis pathways. It is formed from acetyl CoA and acetoacetyl CoA by HMG-CoA synthase. The research of Minor J. Coon and Bimal Kumar Bachhawat in the 1950s at University of Illinois led to its discovery.

Enoyl-CoA hydratase (ECH) or crotonase is an enzyme that hydrates the double bond between the second and third carbons on 2-trans/cis-enoyl-CoA:
Methylcrotonyl CoA carboxylase (MCC) is a biotin-requiring enzyme located in the mitochondria. MCC uses bicarbonate as a carboxyl group source to catalyze the carboxylation of a carbon adjacent to a carbonyl group performing the fourth step in processing leucine, an essential amino acid.
D-Bifunctional protein deficiency is an autosomal recessive peroxisomal fatty acid oxidation disorder. Peroxisomal disorders are usually caused by a combination of peroxisomal assembly defects or by deficiencies of specific peroxisomal enzymes. The peroxisome is an organelle in the cell similar to the lysosome that functions to detoxify the cell. Peroxisomes contain many different enzymes, such as catalase, and their main function is to neutralize free radicals and detoxify drugs. For this reason peroxisomes are ubiquitous in the liver and kidney. D-BP deficiency is the most severe peroxisomal disorder, often resembling Zellweger syndrome.

Adenylosuccinate lyase is an enzyme that in humans is encoded by the ADSL gene.

Isovaleryl-coenzyme A, also known as isovaleryl-CoA, is an intermediate in the metabolism of branched-chain amino acids.

3-Methylglutaconyl-CoA (MG-CoA), also known as β-methylglutaconyl-CoA, is an intermediate in the metabolism of leucine. It is metabolized into HMG-CoA.

3-Methylglutaconyl-CoA hydratase, also known as MG-CoA hydratase and AUH, is an enzyme encoded by the AUH gene on chromosome 19. It is a member of the enoyl-CoA hydratase/isomerase superfamily, but it is the only member of that family that is able to bind to RNA. Not only does it bind to RNA, AUH has also been observed to be involved in the metabolic enzymatic activity, making it a dual-role protein. Mutations of this gene have been found to cause a disease called 3-Methylglutaconic Acuduria Type 1.

In enzymology, an isovaleryl-CoA dehydrogenase is an enzyme that catalyzes the chemical reaction
The discovery of HMG-CoA (3-hydroxy-3-methylglutaryl-CoA) reductase inhibitors, called statins, was a breakthrough in the prevention of hypercholesterolemia and related diseases. Hypercholesterolemia is considered to be one of the major risk factors for atherosclerosis which often leads to cardiovascular, cerebrovascular and peripheral vascular diseases. The statins inhibit cholesterol synthesis in the body and that leads to reduction in blood cholesterol levels, which is thought to reduce the risk of atherosclerosis and diseases caused by it.

Diphosphomevalonate decarboxylase (EC 4.1.1.33), most commonly referred to in scientific literature as mevalonate diphosphate decarboxylase, is an enzyme that catalyzes the chemical reaction

In molecular biology, hydroxymethylglutaryl-CoA synthase or HMG-CoA synthase EC 2.3.3.10 is an enzyme which catalyzes the reaction in which acetyl-CoA condenses with acetoacetyl-CoA to form 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA). This reaction comprises the second step in the mevalonate-dependent isoprenoid biosynthesis pathway. HMG-CoA is an intermediate in both cholesterol synthesis and ketogenesis. This reaction is overactivated in patients with diabetes mellitus type 1 if left untreated, due to prolonged insulin deficiency and the exhaustion of substrates for gluconeogenesis and the TCA cycle, notably oxaloacetate. This results in shunting of excess acetyl-CoA into the ketone synthesis pathway via HMG-CoA, leading to the development of diabetic ketoacidosis.

3-hydroxy-3-methylglutaryl-CoA synthase 2 (mitochondrial) is an enzyme in humans that is encoded by the HMGCS2 gene.

β-Hydroxy β-methylbutyryl-coenzyme A (HMB-CoA), also known as 3-hydroxyisovaleryl-CoA, is a metabolite of L-leucine that is produced in the human body. Its immediate precursors are β-hydroxy β-methylbutyric acid (HMB) and β-methylcrotonoyl-CoA (MC-CoA). It can be metabolized into HMB, MC-CoA, and HMG-CoA in humans.

