The first drug introduced in this class was ticlopidine but due to adverse effects it is not much used today. Ticlopidine, clopidogrel and prasugrel (Efient) are all thienopyridines that cause irreversible inhibition of P2Y12 receptor. They are all prodrugs which need to be converted to an active metabolite in-vivo to inhibit the P2Y12 receptor. On the other hand, novel drugs like ticagrelor (Brilinta®) and cangrelor (Kengrexal®) are non-thienopyridines and reversibly inhibit P2Y12 meaning they act directly on the receptor without the requirement of metabolic activation and display faster onset and offset of action.[1][2][3][4]
These drugs are frequently administered in combination with aspirin (acetylsalicylic acid) to enhance platelet inhibition especially in patients with ACS or undergoing percutaneous coronary intervention (PCI).[5]
History
Before the time of ADP inhibitors the only antiplatelet agent on the market to treat antithrombotic events was aspirin. However, because of recurrent ischemic events in high risk patients there was a basis for development of antiplatelet drugs to target other important signaling pathways.[5][6]
The history of ADP inhibitors started in the year 1972 when researchers were searching for drugs similar to the anti-inflammatory agent tinoridine, a thienopyridine with anti-inflammatory and analgesic effects that had been published two years before.[7] Based on the knowledge of thienopyridine's chemistry a significant number of derivatives of thienopyridines were synthesized. The derivatives were tested in-vivo and ex-vivo in mice and rats but the results of the tests didn't demonstrate any anti-inflammatory or analgesic effects at all but instead they displayed unexpected antiplatelet and antithrombotic effects. At that time it was very uncommon to screen for new antiplatelet agents, as the connection between platelet aggregation, thrombosis and cardiovascular incidents was disputed. However the most active derivative, ticlopidine, was selected for further development.[7] Ticlopidine was the first-generation thienopyridine that enhanced platelet inhibition and thus used for treating in actue coronary syndrome and other cardiovascular diseases. Due to reported severe adverse effects of ticlopidine second and third-thienopyridines, clopidogrel and prasugrel, were developed.[5]
When ticlopidine and clopidogrel were first brought to the market, ticlodipine in 1978 and clopidogrel in 1998, the mechanism of action of these two major antithrombotic drugs was not fully understood. What had been shown was that they were potent inhibitors of ADP-induced platelet aggregation, but the P2Y12 receptor had not been identified. Furthermore, it was clear that ticlopidine and clopidogrel were prodrugs which means they are inactive in-vitro and therefore need metabolism to be activated in-vivo. It wasn't until the year 2000 when the active metabolites of these drugs were isolated and characterized. About one year later the P2Y12 platelet receptor for ADP was identified. Following these discoveries the active metabolites and the enzymes responsible for their formation were progressively identified. Thus it was more than 30 years later after the discovery of ticlopidine, and more than 10 years later after the discovery of clopidogrel the mechanism of action of these two drugs was explained.[7]
Development
Thienopyridines
Ticlopidine
The first P2Y12 inhibitors were of the thienopyridine family. They are indirect antagonists, which block the ADP-induced platelet aggregation and activation. The first drug of this class was ticlopidine and was discovered in 1972 at Porcor (now Sanofi). It was discovered while screening for a new anti-inflammatory drug based on tinoridine. It was screened with a phenotypic screening approach, tested both in vivo and ex vivorodent models. It showed a high antiplatelet activity.[1]
Ticlopidine had good promises and was selected for clinical trials.[1] It was marketed in France in 1978 and went global 1991 when it reached US market[7] for the primary and secondary prevention of stroke.[1]
The search for another thienopyridine analog with a better activity/toxicity ratio in animals started as soon as ticlopidine went into preclinical trials. It became more urgent to find a new analog after reports of patients having severe hematological disorders due to ticlopidine.[7]
Clopidogrel
Clopidogrel, a second generation thienopyridine, started in preclinical trials in 1987 and reached global market in 1998. Its mechanism of action and of its precursor ticlopidine was still unknown. The only things that were known were that they were prodrugs as they didn't show any activity in vitro, that they affect platelets irreversibly because of their long duration of action and the active metabolite was chemically and biologically unstable. It wasn't until 2000 that the active metabolite was discovered and its platelet target was discovered one year later, the P2Y12 receptor of ADP.[7]
Although clopidogrel had better activity/toxicity ratio than ticlopidine there were still problems with its activity as 30% of patients have clopidogrel resistance. The major factor in clopidogrel resistance is CYP2C19 polymorphism, which occurs in 30-55% patients. This led to loss of functions of the enzyme which led to poor conversion of clopidogrel into its active metabolite.[1] The metabolite itself is very unstable and can therefore not be stored as a part of a chemical library.[7] This led to the development of a compound which relies less on CYP-mediated metabolism, prasugrel.[1]
Prasugrel
Prasugrel, third generation thienopyridine was brought to the market in 2009 by the pharmaceutical companies, Daiichi Sankyo/Eli Lilly.[1] Prasugrel, like its precursors is a pro-drug but its metabolism starts in the intestines where it is metabolized by esterase into a thiolactone, this inactive intermediate then undergoes CYP-mediated ring opening, mainly by CYP3A4 and CYP2B6 to the active metabolite. Thus, prasugrel is not subjected to clopidogrel resistance.[8][1]
New generation ADP receptor inhibitors
Ticagrelor
The focus went towards finding a P2Y12 inhibitor that is not a metabolite and with faster onset of action. It was known that ATP competitively antagonizes ADP-induced platelet aggregation, however ATP is very unstable. The attention went to create ATP analogues with higher potency and stability. These analogues had very short half-life due to retention of the triphosphate groups and thus needed to be given IV. Modification of these analogues led to the discovery of ticagrelor, a selective and stable non-phosphate P2Y12 receptor antagonist.[9] Ticagrelor belongs to the class of cyclopentyl-triazolopyrimidine (CPTP).[10] Ticagrelor came to the market in 2010 in Europe,[11] and 2011 in USA.[12]
Cangrelor
Cangrelor another ATP analogue like ticagrelor, is stable to enzymatic degradation. It has a fast onset of action as it is not broken down into an active metabolite like thienopyridines.[13]AstraZeneca got exclusive license for cangrelor in December 2003. In 2009 the sponsor for the phase 3 trial pulled out, where cangrelor was being tested against placebo. Cangrelor development was halted for a time when the sponsor pulled out after the interim analysis review committee (IARC) decided that the trial wouldn't show the "persuasive" clinical efficacy that is need for regulatory approval.[14] However Champion phoenix trial (sponsored by the biopharmaceutical company, The Medicines Company), a double-dummy, double-blind placebo-controlled trial where 11,145 patients who were undergoing rather urgent or elective PCI were randomly assigned to receive cangrelor or clopidogrel before PCI showed that cangrelor significantly reduced the rate of ischemic events during PCI. Cangrelor lowered stent thrombosis development more than clopidogrel. These findings were published in 2013.[15] Cangrelor got FDA approval in June 2015 as an antiplatelet drug for intravenous application.[16]
Mechanism of action
The molecular target of the active metabolite of ADP receptor inhibitors is the P2Y12 receptor.[17] P2Y12 receptor is a G-coupled receptor and is activated by adenosine diphosphate. ADP binds to the P2Y12 receptor that leads to inhibition of adenyl cyclase and thereby decreases the intracellular levels of cAMP. This reduction of cAMP reduces phosphorylation of vasodilator stimulated phosphoprotein that leads to the activation of the glycoprotein IIb/IIIa receptors.[18] Activation of the glycoprotein IIb/IIIa receptors increases thromboxane production and prolonged platelet aggregation.[19] Ticlopidine, clopidogrel and prasugrel are thienopyridine prodrugs that are irreversible platelet inhibitors of the P2Y12 receptor. Cangrelor and ticagrelor are direct –acting P2Y12 inhibitors that change the conformation of the P2Y12 receptor and therefore, results in reversible platelet inhibition of the receptor. Thienopyridines are metabolized in the liver and the intestinal to active metabolites.[20]
P2Y12 antagonists and how they bind to the receptor.
Metabolism
Ticlopidine is a prodrug and is metabolized by at least five main pathways. There is one active metabolite that has been identified and shown to have antiplatelet activity. This active metabolite is formed by a CYP-dependent pathway. CYP2C19 and CYP2B6 are enzymes suggested to contribute to the metabolic transformation of ticlopidine to the thiolactone intermediate, 2-oxo-ticlopidine in the liver. The thiolactone intermediate is then converted to ticlopidine active metabolite via CYP oxidation where oxidation activation occurs. However the CYP enzymes that are involved in this pathway are unknown.[20] In the formation of the active metabolite additional metabolites have been identified and they are dihydrothienopyridinium (M5) and thienodihydropyridinium metabolites (M6). These metabolites may be responsible for the toxic side effects of ticlopidine.[18]
Metabolism of ticlopidine, clopidogrel and prasugrel to an active metabolite.
Clopidogrel is a prodrug that is metabolized by two pathways. In one of the pathway most of the dose of clopidogrel (85%) is hydrolyzed by esterases to an inactive carboxylic acid derivate and rapidly cleared via glucoridination followed by renal excretion. The other pathway of clopidogrel requires a two step hepatic CYP450 metabolic activation to produce the active metabolite that inhibits the P2Y12 receptor. CYP1A2, CYP3A4, CYP3A5 and CYP2C19 are considered to be the main enzymes involved in clopidogrel metabolism.[19] First clopidogrel is metabolised into 2-oxo-clopidogrel, which in turn is hydrolyzed to the active metabolism which is a thiol. The thiol forms a disulfide bridge to a cysteine in P2Y12 receptor and thus binds irreversibly to the P2Y12 receptor. Clopidogrel is suggested to bind covalently to CYS17 or CYS270 of the P2Y12 receptor and therefore blocking the binding of the agonist.[18] Some clopidogrel users have defective CYP2C19 activity and therefore poor CYP2C19 metabolism that leads to the risk of reduced activity of clopidogrel. This is because the prodrug does not metabolized to the active drug. Drugs that are CYP2C19 inhibitors can interact with clopidogrel and reduce clopidogrel activity. All proton pump inhibitors except for rabeprazole and pantoprazole are metabolized by the hepatic CYP450 enzyme and therefore, may interact with the metabolism of clopidogrel. Omeprazole is considered to have higher potential for drug-drug interaction than other protein pump inhibitors because it is a CYP2C19 inhibitor.[17]
Prasugrel is a third generation thienopyridine and a prodrug. Unlike ticlopidine and clopidogrel, the activation of prasugrel involves hydrolysis to an intermediate thiolactone, which is then converted to the active metabolite in a single CYP-dependent step. The active metabolite, which is R-138727 (thiol intermediate), either binds irreversibly to the P2Y12 receptor or is metabolic methylated to an inactive metabolite. R-138727 is metabolically inactivated via S-methylation. Prasugrel is not metabolized by CYP2C19 like clopidogrel and genetic CYP variants do not have a significant influence on the active metabolites of prasugrel. Prasugrel has more rapid onset of action and greater receptor blocking with the active metabolite than clopidogrel.[18]
Ticagrelor was the first reversible inhibitor of the P2Y12 receptor, active after oral administration. Ticagrelor is orally active without the need for any metabolic activation. It is rapidly absorbed and undergoes enzymatic degradation to at least one active metabolite which is almost as potent as its parent compound. Ticagrelor has improved pharmacokinetic and pharmacodynamic profiles compared to currently available drugs for treating ACS. Moreover, CYP2C19 genotypes that are known to influence the effect of clopidogrel did not influence the effect of ticagrelor.[19]
Cangrelor is the first reversible P2Y12 inhibitor that can by administered intravenously and has a very fast onset and offset, which may provide advantages over current drugs and allow cangrelor to overcome these limitations of oral P2Y12 inhibitor. This makes it an appealing option for antiplatelet therapy, especially for patients who are unable to take oral drugs (e.g. patient who are unconscious, vomiting or sedated). Like ticagrelor it does not require metabolic conversion to be active and therefore it can directly inhibit the P2Y12 receptor.[21]
Drug design
Structure-activity relationship (SAR)
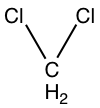
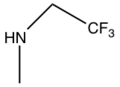
Prasugrel was developed with the metabolism in mind. This was done by replacing the ester group with metabolically stable ketone (group R1). Also the addition of ester group at the thiophene 2-position (group R2) shifted the first step of activation from CYP2C19 to esterases and therefore is prasugrel not metabolized by CYP2C19 like clopidogrel.[22]
SAR for clopidogrel and prasugrel, group R1, R2 and R3 are different.
Compound
R1
R2
R3
Clopidogrel
C
Cl
Prasugrel
F
Cangrelor and ticagrelor are new classes of reversible P2Y12 receptors that have been developed to target the issues of safety (bleeding) and non-responders which the thienopyridines have. Natural ligands like adenosine triphosphate (ATP) were shown to inhibit platelet aggregation and has been identified as a weak antagonist. Cangrelor and ticagrelor are nucleotide analogues that have a chemical structure that resembles adenosine triphosphate (ATP).[21] ATP can be metabolised in cardiovascular tissues to pro-aggregatory ADP, AMP or adenosine. Replacement of anhydride oxygen between phosphorus β (Pβ) and phosphorus γ (Pγ) with di-Chloro or di-Fluoro-methylene leads to a compound that is equipotent to ATP and have a similar pKa. Because of this replacement the metabolism of ATP to pro-aggregatory can be avoided. By adding S-propyl at the chainlength it had a major impact on the activity. Addition of monosubstituted alkyl amine at the C4 position of the adenosine led to a tenfold increase in activity and also the length of the alkylamine substituent correlated with offset of effect. By adding methylsulfanylethylamino group at the C4 position and trifluoropropylsulfanyl at the chainlength leads to the formation of the drug cangrelor that has enhanced activity. Cangrelor has a 78% mean recovery of ADP induced platelet aggregation in rat after 20 minutes comparison to compound 1C which has a less than 10% recovery.[22]
SAR for cangrelor and ticagrelor, group R1, R2 and R3 are different.
Compound
pIC50
R1
R2
R3
ATP
3.6
O
C
C-NH2
1A
3.5
C
C-NH2
1B
8.6
C-NH2
1C
9.1
Cangrelor
9.4
To meet the need of the reversible orally drug ticagrelor the phosphate chain of cangrelor was replaced with an aspartic acid, resulting in 300 fold reduction in potency. The potency of ticagrelor was brought back to the same level as cangrelor by changing the purine with triazolopyrimidine. The sugar ribose unit was also replaced with a cyclopentyl group to avoid possible instability of the glycosidic bond. The group at the left hand side of the structure was replaced with the sidecain R1. The neutral sidechain R1=CONH2 and R1=CH2OH were accepted with slight loss of affinity and the metabolism had shifted from biliary to hepatic metabolism. Because of this, in vitro hepatic microsomal assays could be used which simplified the optimization of pharmacokinetic properties. Addition of phenyl cyclopropylamine substituent in the 5 position gave high affinities. From this the first compound was found to have measurable oral bioavailability in rats (R1 =CH2OH). Variation of R2 had minor impact on affinity that allowed introduction of groups to improve pharmacokinetic properties, for example R1=OCH2CH2OH. The introduction of fluorines at the phenyl ring and at the end of the thioether alkylchain leads to further improved metabolic stability. By replacing the fluorines at the thioether alkylchain back to S-propyl it leads to the formulation of ticagrelor.[22]
SAR, group R1, R2, R3, R4 and R5
Compound
pIC50
R1
R2
R3
R4
R5
ATP
3.6
C
O
C-NH2
C
2A
7.0
C
O
2B
9.3
N
C
2C
COOH=9.3 CONH2 =7.7
CH2OH=7.1
COOH, CONH2 or
CH2OH
N
C
2D
COOH=9.6
CONH2=8.8
CH2OH=8.3
H=8.6
OCH2CH2OH = 8.5
COOH, CONH2, CH2OH, H or
OCH2CH2OH
N
C
2E
9.2
OCH2CH2OH
N
C
Ticagrelor
8.3
OCH2CH2OH
N
C
Clinical use
Activation of platelets and the subsequent aggregation of platelets has a crucial role maintaining normal haemostasis. Disturbance in this system can lead to cerebrovascular, cardiovascular and peripheral vascular diseases where it can lead to a stroke, unstable angina and myocardial infarction. When a vessel is damaged ADP is released from damaged cells and activated platelets, inducing further platelet aggregation.[23][24]
The second generation thienopyridine P2Y12 receptor blocker clopidogrel is an effective antiplatelet agent useful for treatment of ischemic cerebrovascular, cardiac and peripheral arterial release.[27] Like other thienopyridine drugs, the drug was often combined with aspirin in clinical use.[23] The clinically approved dosage of clopidogrel is a 300-mg loading dosePO and a 75-mg a day maintenance dose PO.[28]
For many years dual treatment with the cyclooxygenase-1 (COX-1) inhibitor aspirin and clopidogrel was routine practice and served as the main antiplatelet agents for the prevention of thrombotic events as they have the capability to powerfully manipulate platelet biology, which plays a central part in thrombosis. However, the use of these agents is still subjected to a number of important limitations such as exposure to increased risk of bleeding, making duration and dosage of clopidogrel of the utmost importance. Furthermore, responsiveness to clopidogrel is not uniform and low response can lead to major adverse cardiovascular events.[29][30]
New generations
The new generation of P2Y12 blockers aimed to address these issues, promising improvement in outcome for patients. These recently developed P2Y12 blockers (ticagrelor, cangrelor, prasugrel and elinogrel) provide a more consistent and stronger inhibition of platelets by more efficiently antagonizing the P2Y12 receptor. However, this more potent platelet inhibition comes at the cost of a higher bleeding risk.[31][29]
Prasugrel, a third generation thienopyridine, is metabolized more efficiently than clopidogrel and ticlopidine in the body and therefore it prevents platelet activation to a greater extent. Studies have shown prasugrel to reduce the risk of stent thrombosis and myocardial infarction to much greater level than clopidogrel.[26] The clinically approved dose of prasugrel is a 60-mg loading dose PO and a 10-mg a day maintenance dose PO.[28]
Ticagrelor is a much more potent inhibitor of platelet aggregation than clopidogrel, however, it is associated with increase of dyspnoea episodes in patients. These episodes can range from mild to moderate severity. The approved clinical dosage of ticagrelor is a 180-mg loading dose PO and a 90-mg a day maintenance dose.[32]
The only parental drug targeting the P2Y12 receptor in clinical use is cangrelor.[33]
Interactions
The CYP2C19 enzyme metabolizes proton pump inhibitors (PPI) as well as clopidogrel. Various reports have stated that there is a negative clopidogrel-omeprazole drug interaction. Some studies have found that clopidogrel activity on platelets was hampered significantly by patients receiving treatment with omeprazole, a proton pump inhibitor (PPI).[34][35] Another study also showed lansoprazole to have hampering effects on clopidogrel activity.[36] However, other studies have shown the intake of the PPI's pantoprazole or esomeprazole not to be associated with impaired response to clopidogrel.[35][37]
In 2009 the United States food and drug administration (FDA) and the European Medicines Agency (EMA) discouraged the combination of clopidogrel and PPI's, especially omeprazole, due to observations made at the time by Initial Cohort Studies. However, newer retrospectivecohort studies have not shown adverse cardiovascular events caused by clopidogrel-PPI interactions. Therefore there is no definite evidence on the drug interaction effect on mortality.[38]
For patients with high risk of gastrointestinal bleeding, the risk outweighs the possible adverse cardiovascular effects. It should therefore be recommended to those patients to combine clopidogrel with less CYP2C19 inhibiting PPI's, such as pantoprazole.[38]
Clopidogrel resistance
The resistance of clopidogrel has emerged through the years and become a great concern for the therapy of patients with ACS or undergoing PCI. Clopidogrel resistance is reported to vary from 4-44% between different populations and ethnic groups. Patients who are exposed to clopidogrel resistance display lower activity of platelet inhibition due to decreased levels of the active metabolite of clopidogrel. This results in series of clinical incidents, e.g. ischemic and thromboembolic complications. These patients are recognized as poor- or non-responders.[39][40]
Clopidogrel is a prodrug that needs a two-step metabolization with the help of enzymes to become an active metabolite. One of the crucial enzymes in clopidogrel metabolism is CYP2C19 which is involved in both steps of the biotransformation. A polymorphism of the enzyme CYP2C19 affects the response to clopidogrel hence decreasing enzymatic activity and therefore reducing the active metabolite of clopidogrel.[40]
The major issue of clopidogrel resistance is the interaction with other drugs, especially the proton pump inhibitor omeprazole. Omeprazole and clopidogrel are metabolized by the same CYP metabolic pathway. Consequently, it has been suggested that the low-responsiveness of clopidogrel, with concomitant use of omeprazole, is caused by the competition of the CYP2C19 enzyme between these two drugs.[40]
Clinical approaches on how to overcome clopidogrel resistance include higher doses of clopidogrel, concomitant use with the phosphodiesterase inhibitorcilostazol or switching to a new antiplatelet agent such as prasugrel and ticagrelor. Still, the major concern with these methods above are increased risk of bleeding. Therefore, the need for a new antiplatelet agent with fast onset of action, less variability in response among individuals and improved safety profile is critical.[39]
Future prospects
The development of ADP inhibitors is constantly advancing and the search for even better P2Y12 antagonists is still ongoing.[1] The cornerstone of secondary prevention of atherothrombotic events in patients with ACS or undergoing PCI is dual antiplatelet therapy with aspirin and clopidogrel. Nevertheless, events of atherothrombosis still occur.[41] The limitations of current antiplatelet drugs contain risk of bleeding and interindividual variability of platelet inhibitory response.[6]
The aim is to determine the optimal therapeutic window to maximize therapeutic benefits while reducing safety concerns like bleeding. Consequently, the major unmet goal of ADP inhibitors is to develop a potent reversible antiplatelet agent with fast onset of action, high levels of antiplatelet activity yet decreased risk of bleedings. This challenging goal together with a once-daily oral profile and optimized target selectivity would possibly be an important breakthrough in this field.[1][39]
Vicagrel is the latest development, as of September 2017, in this sector. A novel acetate analog of clopidogrel that is expected to achieve improved antiplatelet efficiency as well as decreased risk of bleeding.[42] Preliminary pharmacokinetics studies of vicagrel have shown a higher bioavailability than in clopidogrel, indicating a much lower therapeutic effective dose for vicagrel. The advantages of vicagrel over clopidogrel are considered to be no drug resistance for CYP2C19 poor metabolizers, decreased dose-related toxicity because of lower effective dose and faster onset of action.[39]
References
1234567891011Martinez, Ana; Gil, Carmen, eds. (2017-07-19). Comprehensive medicinal chemistry. Volume 1, General perspective — the future of drug discovery (3rded.). Amsterdam, the Netherlands. ISBN9780128032008. OCLC989872324.{{cite book}}: CS1 maint: location missing publisher (link)[pageneeded]
123K., Chopra, H. (2012-12-30). Textbook of cardiology: a clinical & historical perspective. Nanda, Navin C. (Navin Chandar), 1937-. New Delhi. ISBN9789350900819. OCLC870265462.{{cite book}}: CS1 maint: location missing publisher (link) CS1 maint: multiple names: authors list (link)[pageneeded]
12Angiolillo, Dominick J.; Luis Ferreiro, José (January 2010). "Platelet Adenosine Diphosphate P2Y12 Receptor Antagonism: Benefits and Limitations of Current Treatment Strategies and Future Directions". Revista Española de Cardiología. 63 (1): 60–76. doi:10.1016/s1885-5857(10)70010-4. PMID20089227. S2CID24637687.
↑Storey, Robert (6 December 2017). "Pharmacology and clinical trials of reversibly-binding P2Y12 inhibitors". Thrombosis and Haemostasis. 105 (S 06): S75–S81. doi:10.1160/THS10-12-0769. PMID21479343. S2CID3261918.
1234Lemke, Thomas L.; Williams, David A., eds. (2013). Foye's principles of medicinal chemistry (7thed.). Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins. ISBN9781451175721. OCLC748675182.[pageneeded]
12Farid, Nagy A.; Kurihara, Atsushi; Wrighton, Steven A. (February 2010). "Metabolism and Disposition of the Thienopyridine Antiplatelet Drugs Ticlopidine, Clopidogrel, and Prasugrel in Humans". The Journal of Clinical Pharmacology. 50 (2): 126–142. doi:10.1177/0091270009343005. PMID19948947. S2CID31554679.
12Angiolillo, Dominick J.; Bates, Eric R.; Bass, Theodore A. (August 2008). "Clinical profile of prasugrel, a novel thienopyridine". American Heart Journal. 156 (2): 16S–22S. doi:10.1016/j.ahj.2008.06.005. PMID18657682.
12Sarafoff, Nikolaus; A. Byrne, Robert; Sibbing, Dirk (23 September 2012). "Clinical Use of Clopidogrel". Current Pharmaceutical Design. 18 (33): 5224–5239. doi:10.2174/138161212803251853. PMID22724411.
↑Tang, Jie; Li, Mu-Peng; Zhou, Hong-Hao; Chen, Xiao-Ping (25 August 2015). "Platelet Inhibition Agents: Current and Future P2Y12 Receptor Antagonists". Current Vascular Pharmacology. 13 (5): 566–577. doi:10.2174/1570161112666141127162209. PMID25440595.
↑Ahmad, Shiraz; F. Storey, Robert (23 September 2012). "Development and Clinical use of Prasugrel and Ticagrelor". Current Pharmaceutical Design. 18 (33): 5240–5260. doi:10.2174/138161212803251989. PMID22724412.
↑Small, David S.; Farid, Nagy A.; Payne, Christopher D.; Weerakkody, Govinda J.; Li, Ying G.; Brandt, John T.; Salazar, Daniel E.; Winters, Kenneth J. (April 2008). "Effects of the Proton Pump Inhibitor Lansoprazole on the Pharmacokinetics and Pharmacodynamics of Prasugrel and Clopidogrel". The Journal of Clinical Pharmacology. 48 (4): 475–484. doi:10.1177/0091270008315310. PMID18303127. S2CID6216572.
↑Siller-Matula, Jolanta M.; Spiel, Alexander O.; Lang, Irene M.; Kreiner, Gerhard; Christ, Guenter; Jilma, Bernd (January 2009). "Effects of pantoprazole and esomeprazole on platelet inhibition by clopidogrel". American Heart Journal. 157 (1): 148.e1–148.e5. doi:10.1016/j.ahj.2008.09.017. PMID19081411.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.