This article needs additional citations for verification .(February 2009) (Learn how and when to remove this template message) |
| Arrhythmogenic cardiomyopathy | |
|---|---|
| Other names | arrhythmogenic right ventricular cardiomyopathy (ARVC), arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C), right ventricular dysplasia |
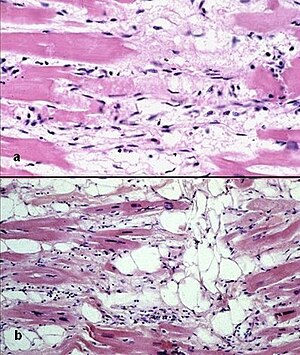 | |
| Photomicrograph of an ACM heart. | |
| Specialty | Cardiology |
Arrhythmogenic cardiomyopathy(ACM), arrhythmogenic right ventricular dysplasia (ARVD), or arrhythmogenic right ventricular cardiomyopathy (ARVC), is an inherited heart disease. [1]
Contents
- Signs and symptoms
- Genetics
- Pathogenesis
- Fatty infiltration
- Fibro-fatty infiltration
- The Role of Exercise
- Ventricular arrhythmias
- Diagnosis
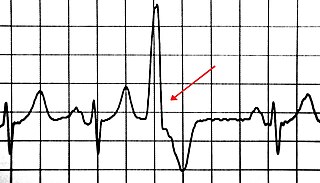
- Electrocardiogram
- Echocardiography
- MRI
- Angiography
- Biopsy
- Genetic testing
- Diagnostic criteria
- Management
- Medications
- Catheter ablation
- Implantable cardioverter-defibrillator
- Heart transplant
- Family screening
- Prognosis
- Epidemiology
- Notable cases
- See also
- References
- External links
ACM is caused by genetic defects of the parts of heart muscle (also called myocardium or cardiac muscle) known as desmosomes, areas on the surface of heart muscle cells which link the cells together. The desmosomes are composed of several proteins, and many of those proteins can have harmful mutations.

A genetic disorder is a genetic problem caused by one or more abnormalities formed in the genome. Most genetic disorders are quite rare and affect one person in every several disease thousands or millions. The earliest known genetic condition in a hominid was in the fossil species Paranthropus robustus, with over a third of individuals displaying Amelogenesis imperfecta.

Cardiac muscle is one of three types of vertebrate muscles, with the other two being skeletal and smooth muscles. It is an involuntary, striated muscle that constitutes the main tissue of the walls of the heart. The myocardium forms a thick middle layer between the outer layer of the heart wall and the inner layer, with blood supplied via the coronary circulation. It is composed of individual heart muscle cells (cardiomyocytes) joined together by intercalated discs, encased by collagen fibres and other substances that form the extracellular matrix.

A desmosome, also known as a macula adherens, is a cell structure specialized for cell-to-cell adhesion. A type of junctional complex, they are localized spot-like adhesions randomly arranged on the lateral sides of plasma membranes. Desmosomes are one of the stronger cell-to-cell adhesion types and are found in tissue that experience intense mechanical stress, such as cardiac muscle tissue, bladder tissue, gastrointestinal mucosa, and epithelia.
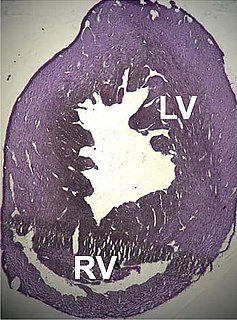
The disease is a type of nonischemic cardiomyopathy that primarily involves the right ventricle, though cases of exclusive left ventricular disease have been reported. It is characterized by hypokinetic areas involving the free wall of the ventricle, with fibrofatty replacement of the myocardium, with associated arrhythmias often originating in the right ventricle. The nomenclature ARVD is currently thought to be inappropriate and misleading as ACM does not involve dysplasia of the ventricular wall. Cases of ACM originating from the left ventricle lead to the abandonment of the name ARVC.

Cardiomyopathy is a group of diseases that affect the heart muscle. Early on there may be few or no symptoms. As the disease worsens, shortness of breath, feeling tired, and swelling of the legs may occur, due to the onset of heart failure. An irregular heart beat and fainting may occur. Those affected are at an increased risk of sudden cardiac death.

A ventricle is one of two large chambers toward the bottom of the heart that collect and expel blood received from an atrium towards the peripheral beds within the body and lungs. The atrium primes the pump.
Hypokinesia refers to decreased bodily movement. One of the two categories of movement disorders, hypokinesia is characterized by a partial or complete loss of muscle movement due to a disruption in the basal ganglia. Patients with hypokinetic disorders like Parkinson's disease experience muscle rigidity and an inability to produce movement. It is also associated with mental health disorders and prolonged inactivity due to illness, amongst other diseases.
ACM can be found in association with diffuse palmoplantar keratoderma, and woolly hair, in an autosomal recessive condition called Naxos disease, because this genetic abnormality can also affect the integrity of the superficial layers of the skin most exposed to pressure stress. [2] :513 [3]
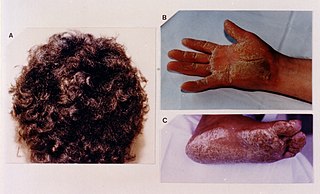
Naxos disease is a cutaneous condition characterized by a palmoplantar keratoderma. The prevalence of the syndrome is up to 1 in every 1000 people in the Greek islands.
ACM is an important cause of ventricular arrhythmias in children and young adults. It is seen predominantly in males, and 30–50% of cases have a familial distribution.