
The cannabinoid receptor 2(CB2), is a G protein-coupled receptor from the cannabinoid receptor family that in humans is encoded by the CNR2 gene. It is closely related to the cannabinoid receptor 1 (CB1), which is largely responsible for the efficacy of endocannabinoid-mediated presynaptic-inhibition, the psychoactive properties of tetrahydrocannabinol (THC), the active agent in cannabis, and other phytocannabinoids. The principal endogenous ligand for the CB2 receptor is 2-Arachidonoylglycerol (2-AG).

JWH-051 is an analgesic drug which is a cannabinoid agonist. Its chemical structure is closely related to that of the potent cannabinoid agonist HU-210, with the only difference being the removal of the hydroxyl group at position 1 of the aromatic ring. It was discovered and named after John W. Huffman.

NESS-0327 is a drug used in scientific research which acts as an extremely potent and selective antagonist of the cannabinoid receptor CB1. It is much more potent an antagonist, and more selective for the CB1 receptor over CB2, than the more commonly used ligand rimonabant, with a Ki at CB1 of 350fM (i.e. 0.00035nM) and a selectivity of over 60,000x for CB1 over CB2. Independently, two other groups have described only modest nanomolar CB1 affinity for this compound (125nM and 18.4nM). Also unlike rimonabant, NESS-0327 does not appear to act as an inverse agonist at higher doses, instead being a purely neutral antagonist which blocks the CB1 receptor but does not produce any physiological effect of its own.

JTE-907 is a drug used in scientific research that acts as a selective CB2 inverse agonist. It has antiinflammatory effects in animal studies, thought to be mediated by an interaction between the CB2 receptor and IgE.
A cannabinoid receptor antagonist, also known simply as a cannabinoid antagonist or as an anticannabinoid, is a type of cannabinoidergic drug that binds to cannabinoid receptors (CBR) and prevents their activation by endocannabinoids. They include antagonists, inverse agonists, and antibodies of CBRs. The discovery of the endocannabinoid system led to the development of CB1 receptor antagonists. The first CBR inverse agonist, rimonabant, was described in 1994. Rimonabant blocks the CB1 receptor selectively and has been shown to decrease food intake and regulate body-weight gain. The prevalence of obesity worldwide is increasing dramatically and has a great impact on public health. The lack of efficient and well-tolerated drugs to cure obesity has led to an increased interest in research and development of CBR antagonists. Cannabidiol (CBD), a naturally occurring cannabinoid and a non-competitive CB1/CB2 receptor antagonist, as well as Δ9-tetrahydrocannabivarin (THCV), a naturally occurring cannabinoid, modulate the effects of THC via direct blockade of cannabinoid CB1 receptors, thus behaving like first-generation CB1 receptor inverse agonists, such as rimonabant. CBD is a very low-affinity CB1 ligand, that can nevertheless affect CB1 receptor activity in vivo in an indirect manner, while THCV is a high-affinity CB1 receptor ligand and potent antagonist in vitro and yet only occasionally produces effects in vivo resulting from CB1 receptor antagonism. THCV has also high affinity for CB2 receptors and signals as a partial agonist, differing from both CBD and rimonabant.

A-834,735 is a drug developed by Abbott Laboratories that acts as a potent cannabinoid receptor full agonist at both the CB1 and CB2 receptors, with a Ki of 12 nM at CB1 and 0.21 nM at CB2. Replacing the aromatic 3-benzoyl or 3-naphthoyl group found in most indole derived cannabinoids with the 3-tetramethylcyclopropylmethanone group of A-834,735 and related compounds imparts significant selectivity for CB2, with most compounds from this group found to be highly selective CB2 agonists with little affinity for CB1. However, low nanomolar CB1 binding affinity is retained with certain heterocyclic 1-position substituents such as (N-methylpiperidin-2-yl)methyl (cf. AM-1220, AM-1248), or the (tetrahydropyran-4-yl)methyl substituent of A-834,735, resulting in compounds that still show significant affinity and efficacy at both receptors despite being CB2 selective overall.

AM-1221 is a drug that acts as a potent and selective agonist for the cannabinoid receptor CB2, with a Ki of 0.28 nM at CB2 and 52.3 nM at the CB1 receptor, giving it around 180 times selectivity for CB2. The 2-methyl and 6-nitro groups on the indole ring both tend to increase CB2 affinity while generally reducing affinity at CB1, explaining the high CB2 selectivity of AM-1221. However, despite this relatively high selectivity for CB2, its CB1 affinity is still too strong to make it useful as a truly selective CB2 agonist, so the related compound AM-1241 is generally preferred for research purposes.

AM-679 (part of the AM cannabinoid series) is a drug that acts as a moderately potent agonist for the cannabinoid receptors, with a Ki of 13.5 nM at CB1 and 49.5 nM at CB2. AM-679 was one of the first 3-(2-iodobenzoyl)indole derivatives that was found to have significant cannabinoid receptor affinity, and while AM-679 itself has only modest affinity for these receptors, it was subsequently used as a base to develop several more specialised cannabinoid ligands that are now widely used in research, including the potent CB1 agonists AM-694 and AM-2233, and the selective CB2 agonist AM-1241. AM-679 was first identified as having been sold as a cannabinoid designer drug in Hungary in 2011, along with another novel compound 1-pentyl-3-(1-adamantoyl)indole.

AM-1235 (1-(5-fluoropentyl)-3-(naphthalen-1-oyl)-6-nitroindole) is a drug that acts as a potent and reasonably selective agonist for the cannabinoid receptor CB1.

AM-1220 is a drug that acts as a potent and moderately selective agonist for the cannabinoid receptor CB1, with around 19 times selectivity for CB1 over the related CB2 receptor. It was originally invented in the early 1990s by a team led by Thomas D'Ambra at Sterling Winthrop, but has subsequently been researched by many others, most notably the team led by Alexandros Makriyannis at the University of Connecticut. The (piperidin-2-yl)methyl side chain of AM-1220 contains a stereocenter, so there are two enantiomers with quite different potency, the (R)-enantiomer having a Ki of 0.27 nM at CB1 while the (S)-enantiomer has a much weaker Ki of 217 nM.

AZ-11713908 is a drug developed by AstraZeneca which is a peripherally selective cannabinoid agonist, acting as a potent agonist at the CB1 receptor and a partial agonist at CB2. It has poor blood–brain barrier penetration, and so while it is an effective analgesic in animal tests, it produces only peripheral effects at low doses, with much weaker symptoms of central effects compared to other cannabinoid drugs such as WIN 55,212-2. Many related benzimidazole-derived cannabinoid ligands are known.
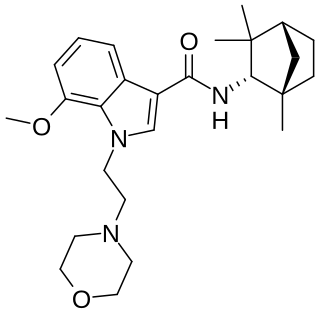
MN-25 (UR-12) is a drug invented by Bristol-Myers Squibb, that acts as a reasonably selective agonist of peripheral cannabinoid receptors. It has moderate affinity for CB2 receptors with a Ki of 11 nM, but 22x lower affinity for the psychoactive CB1 receptors with a Ki of 245 nM. The indole 2-methyl derivative has the ratio of affinities reversed however, with a Ki of 8 nM at CB1 and 29 nM at CB2, which contrasts with the usual trend of 2-methyl derivatives having increased selectivity for CB2 (cf. JWH-018 vs JWH-007, JWH-081 vs JWH-098).

MDA-19 (also known as BZO-HEXOXIZID) is a drug that acts as a potent and selective agonist for the cannabinoid receptor CB2, with reasonable selectivity over the psychoactive CB1 receptor, though with some variation between species. In animal studies it was effective for the treatment of neuropathic pain, but did not affect rat locomotor activity in that specific study. The pharmacology of MDA-19 in rat cannabinoid receptors have been demonstrated to function differently than human cannabinoid receptors with MDA-19 binding to human CB1 receptors 6.9× higher than rat CB1 receptors.

UR-144 (TMCP-018, KM-X1, MN-001, YX-17) is a drug invented by Abbott Laboratories, that acts as a selective full agonist of the peripheral cannabinoid receptor CB2, but with much lower affinity for the psychoactive CB1 receptor.

CBS-0550 is a drug developed by Taisho Pharmaceutical, which acts as a potent and selective cannabinoid CB2 receptor agonist, with 1400x selectivity for CB2 over the related CB1 receptor. Unlike most cannabinoid agonists, CBS-0550 has good solubility in water, and in animal studies it was found to produce analgesic and anti-hyperalgesic effects. A number of related compounds have been developed with similar properties.

S-444,823 is a drug developed by Shionogi which is a cannabinoid agonist. It was developed as an antipruritic, and has moderate selectivity for the CB2 subtype, having a CB2 affinity of 18nM, and 32x selectivity over the CB1 receptor. In animal studies it showed analgesic effects and strongly reduced itching responses, but without producing side effects such as sedation and catalepsy that are seen with centrally acting CB1 agonists.
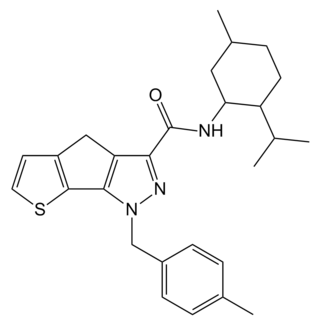
NESS-040C5 is a potent cannabinoid agonist which was developed for the treatment of glaucoma. It has reasonable selectivity for the CB2 receptor subtype, having a CB2 affinity of 0.4nM, and 25x selectivity over the related CB1 receptor.

QMPSB is an arylsulfonamide-based synthetic cannabinoid that has been sold as a designer drug.
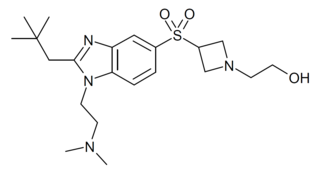
RQ-00202730 is a benzimidazole derived drug that acts as a potent and highly selective agonist for the CB2 cannabinoid receptor, with a Ki value of 19nM at CB2 and more than 4000x selectivity over CB1, though it also shows some activity as an antagonist of the unrelated 5-HT2B serotonin receptor. It has analgesic and antiinflammatory effects in animal studies, and was developed for the treatment of irritable bowel syndrome, but was ultimately discontinued from development following disappointing results in Phase II clinical trials.
