 | |
 | |
| Clinical data | |
|---|---|
| Trade names | Aerius, others [1] |
| Other names | Descarboethoxyloratadine [2] |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a602002 |
| License data | |
| Pregnancy category |
|
| Routes of administration | By mouth |
| Drug class | Second-generation antihistamine |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | Rapidly absorbed |
| Protein binding | 83–87% |
| Metabolism | UGT2B10, CYP2C8 |
| Metabolites | 3-Hydroxydesloratadine |
| Onset of action | within 1 hour |
| Elimination half-life | 27 hours, 33.7 hours in elderly patients [3] |
| Duration of action | up to 24 hours |
| Excretion | 40% as conjugated metabolites into urine Similar amount into the feces |
| Identifiers | |
| |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.166.554 |
| Chemical and physical data | |
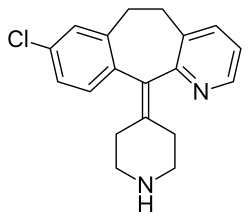
| Formula | C19H19ClN2 |
| Molar mass | 310.83 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
| (verify) | |
Desloratadine, sold under the brand name Aerius among others, is a tricyclic H1 inverse agonist that is used to treat allergies. It is the major active metabolite of loratadine.
Contents
- Medical uses
- Side effects
- Interactions
- Pharmacology
- Pharmacodynamics
- Pharmacokinetics
- Pharmacogenomics
- References
- Further reading
It was patented in 1984 and came into medical use in 2001. [8] It was brought to the market in the US by Schering Corporation, later named Schering-Plough. [3]
