In organic chemistry, an alkyne is an unsaturated hydrocarbon containing at least one carbon—carbon triple bond. The simplest acyclic alkynes with only one triple bond and no other functional groups form a homologous series with the general chemical formula CnH2n−2. Alkynes are traditionally known as acetylenes, although the name acetylene also refers specifically to C2H2, known formally as ethyne using IUPAC nomenclature. Like other hydrocarbons, alkynes are generally hydrophobic.
The following outline is provided as an overview of and topical guide to organic chemistry:
The Sonogashira reaction is a cross-coupling reaction used in organic synthesis to form carbon–carbon bonds. It employs a palladium catalyst as well as copper co-catalyst to form a carbon–carbon bond between a terminal alkyne and an aryl or vinyl halide.
The 1,3-dipolar cycloaddition is a chemical reaction between a 1,3-dipole and a dipolarophile to form a five-membered ring. The earliest 1,3-dipolar cycloadditions were described in the late 19th century to the early 20th century, following the discovery of 1,3-dipoles. Mechanistic investigation and synthetic application were established in the 1960s, primarily through the work of Rolf Huisgen. Hence, the reaction is sometimes referred to as the Huisgen cycloaddition. 1,3-dipolar cycloaddition is an important route to the regio- and stereoselective synthesis of five-membered heterocycles and their ring-opened acyclic derivatives. The dipolarophile is typically an alkene or alkyne, but can be other pi systems. When the dipolarophile is an alkyne, aromatic rings are generally produced.
In organic chemistry, arynes and benzynes are a class of highly reactive chemical species derived from an aromatic ring by removal of two substituents. Arynes are examples of didehydroarenes, although 1,3- and 1,4-didehydroarenes are also known. Arynes are examples of alkynes under high strain.
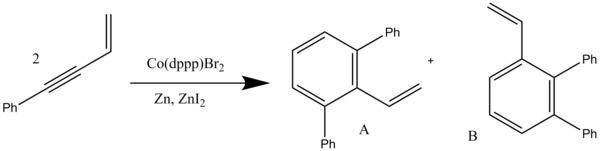
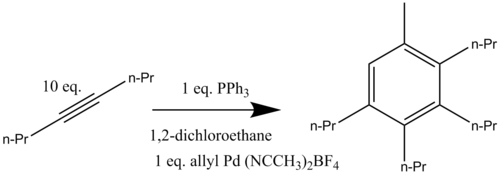
An alkyne trimerisation is a [2+2+2] cycloaddition reaction in which three alkyne units react to form a benzene ring. The reaction requires a metal catalyst. The process is of historic interest as well as being applicable to organic synthesis. Being a cycloaddition reaction, it has high atom economy. Many variations have been developed, including cyclisation of mixtures of alkynes and alkenes as well as alkynes and nitriles.
The Larock indole synthesis is a heteroannulation reaction that uses palladium as a catalyst to synthesize indoles from an ortho-iodoaniline and a disubstituted alkyne. It is also known as Larock heteroannulation. The reaction is extremely versatile and can be used to produce varying types of indoles. Larock indole synthesis was first proposed by Richard C. Larock in 1991 at Iowa State University.

The Povarov reaction is an organic reaction described as a formal cycloaddition between an aromatic imine and an alkene. The imine in this organic reaction is a condensation reaction product from an aniline type compound and a benzaldehyde type compound. The alkene must be electron rich which means that functional groups attached to the alkene must be able to donate electrons. Such alkenes are enol ethers and enamines. The reaction product in the original Povarov reaction is a quinoline. Because the reactions can be carried out with the three components premixed in one reactor it is an example of a multi-component reaction.
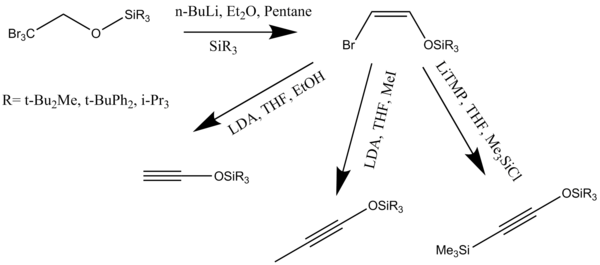
In organic chemistry, a cycloalkyne is the cyclic analog of an alkyne. A cycloalkyne consists of a closed ring of carbon atoms containing one or more triple bonds. Cycloalkynes have a general formula CnH2n−4. Because of the linear nature of the C−C≡C−C alkyne unit, cycloalkynes can be highly strained and can only exist when the number of carbon atoms in the ring is great enough to provide the flexibility necessary to accommodate this geometry. Large alkyne-containing carbocycles may be virtually unstrained, while the smallest constituents of this class of molecules may experience so much strain that they have yet to be observed experimentally. Cyclooctyne is the smallest cycloalkyne capable of being isolated and stored as a stable compound. Despite this, smaller cycloalkynes can be produced and trapped through reactions with other organic molecules or through complexation to transition metals.

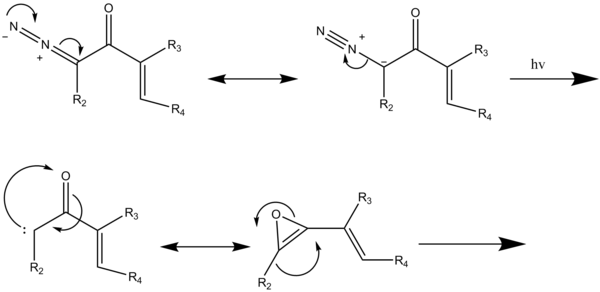
The Wolff rearrangement is a reaction in organic chemistry in which an α-diazocarbonyl compound is converted into a ketene by loss of dinitrogen with accompanying 1,2-rearrangement. The Wolff rearrangement yields a ketene as an intermediate product, which can undergo nucleophilic attack with weakly acidic nucleophiles such as water, alcohols, and amines, to generate carboxylic acid derivatives or undergo [2+2] cycloaddition reactions to form four-membered rings. The mechanism of the Wolff rearrangement has been the subject of debate since its first use. No single mechanism sufficiently describes the reaction, and there are often competing concerted and carbene-mediated pathways; for simplicity, only the textbook, concerted mechanism is shown below. The reaction was discovered by Ludwig Wolff in 1902. The Wolff rearrangement has great synthetic utility due to the accessibility of α-diazocarbonyl compounds, variety of reactions from the ketene intermediate, and stereochemical retention of the migrating group. However, the Wolff rearrangement has limitations due to the highly reactive nature of α-diazocarbonyl compounds, which can undergo a variety of competing reactions.
The Wulff–Dötz reaction (also known as the Dötz reaction or the benzannulation reaction of the Fischer carbene complexes) is the chemical reaction of an aromatic or vinylic alkoxy pentacarbonyl chromium carbene complex with an alkyne and carbon monoxide to give a Cr(CO)3-coordinated substituted phenol. Several reviews have been published. It is named after the German chemist Karl Heinz Dötz (b. 1943) and the American chemist William D. Wulff (b. 1949) at Michigan State University. The reaction was first discovered by Karl Dötz and was extensively developed by his group and W. Wulff's group. They subsequently share the name of the reaction.

In organic chemistry, annulation is a chemical reaction in which a new ring is constructed on a molecule.
The nitrone-olefin (3+2) cycloaddition reaction is the combination of a nitrone with an alkene or alkyne to generate an isoxazoline or isoxazolidine via a (3+2) cycloaddition process. This reaction is a 1,3-dipolar cycloaddition, in which the nitrone acts as the 1,3-dipole, and the alkene or alkyne as the dipolarophile.
The Buchner ring expansion is a two-step organic C-C bond forming reaction used to access 7-membered rings. The first step involves formation of a carbene from ethyl diazoacetate, which cyclopropanates an aromatic ring. The ring expansion occurs in the second step, with an electrocyclic reaction opening the cyclopropane ring to form the 7-membered ring.
Decarboxylative cross coupling reactions are chemical reactions in which a carboxylic acid is reacted with an organic halide to form a new carbon-carbon bond, concomitant with loss of CO2. Aryl and alkyl halides participate. Metal catalyst, base, and oxidant are required.
In organic chemistry, the hexadehydro-Diels–Alder (HDDA) reaction is an organic chemical reaction between a diyne and an alkyne to form a reactive benzyne species, via a [4+2] cycloaddition reaction. This benzyne intermediate then reacts with a suitable trapping agent to form a substituted aromatic product. This reaction is a derivative of the established Diels–Alder reaction and proceeds via a similar [4+2] cycloaddition mechanism. The HDDA reaction is particularly effective for forming heavily functionalized aromatic systems and multiple ring systems in one synthetic step.
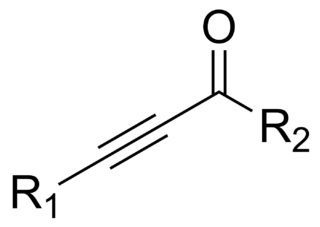
In organic chemistry, an ynone is an organic compound containing a ketone functional group and a C≡C triple bond. Many ynones are α,β-ynones, where the carbonyl and alkyne groups are conjugated. Capillin is a naturally occurring example. Some ynones are not conjugated.

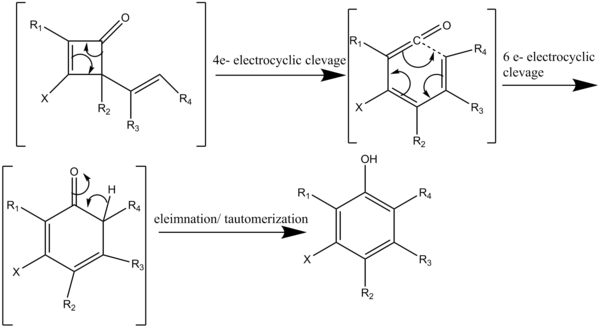
Rick L. Danheiser is an American organic chemist and is the Arthur C. Cope Professor of Chemistry at the Massachusetts Institute of Technology and chair of the MIT faculty. His research involves the invention of new methods for the synthesis of complex organic compounds. Danheiser is known for the Danheiser annulation and Danheiser benzannulation reactions.

In chemistry, a phenol ether (or aromatic ether) is an organic compound derived from phenol (C6H5OH), where the hydroxyl (-OH) group is substituted with an alkoxy (-OR) group. Usually phenol ethers are synthesized through the condensation of phenol and an organic alcohol; however, other known reactions regarding the synthesis of ethers can be applied to phenol ethers as well. Anisole (C6H5OCH3) is the simplest phenol ether, and is a versatile precursor for perfumes and pharmaceuticals. Vanillin and ethylvanillin are phenol ether derivatives commonly utilized in vanilla flavorings and fragrances, while diphenyl ether is commonly used as a synthetic geranium fragrance. Phenol ethers are part of the chemical structure of a variety of medications, including quinine, an antimalarial drug, and dextromethorphan, an over-the-counter cough suppressant.
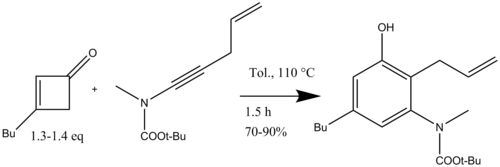
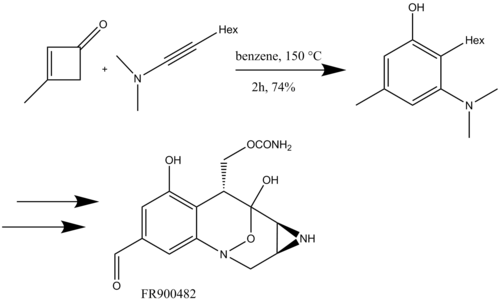
In organic chemistry, the Conia-ene reaction is an intramolecular cyclization reaction between an enolizable carbonyl such as an ester or ketone and an alkyne or alkene, giving a cyclic product with a new carbon-carbon bond. As initially reported by J. M. Conia and P. Le Perchec, the Conia-ene reaction is a heteroatom analog of the ene reaction that uses an enol as the ene component. Like other pericyclic reactions, the original Conia-ene reaction required high temperatures to proceed, limiting its wider application. However, subsequent improvements, particularly in metal catalysis, have led to significant expansion of reaction scope. Consequently, various forms of the Conia-ene reaction have been employed in the synthesis of complex molecules and natural products.