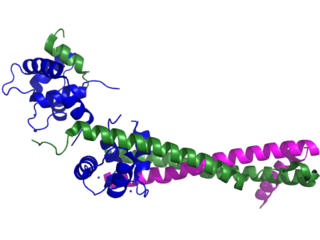
Troponin, or the troponin complex, is a complex of three regulatory proteins that are integral to muscle contraction in skeletal muscle and cardiac muscle, but not smooth muscle. Measurements of cardiac-specific troponins I and T are extensively used as diagnostic and prognostic indicators in the management of myocardial infarction and acute coronary syndrome. Blood troponin levels may be used as a diagnostic marker for stroke, although the sensitivity of this measurement is low.

Titin, also known as connectin, is a protein that in humans is encoded by the TTN gene. Titin is a giant protein, greater than 1 µm in length, that functions as a molecular spring which is responsible for the passive elasticity of muscle. It comprises 244 individually folded protein domains connected by unstructured peptide sequences. These domains unfold when the protein is stretched and refold when the tension is removed.

Tropomyosin is a two-stranded alpha-helical, coiled coil protein found in actin-based cytoskeletons.
Nemaline myopathy is a congenital, often hereditary neuromuscular disorder with many symptoms that can occur such as muscle weakness, hypoventilation, swallowing dysfunction, and impaired speech ability. The severity of these symptoms varies and can change throughout one's life to some extent. The prevalence is estimated at 1 in 50,000 live births. It is the most common non-dystrophic myopathy.
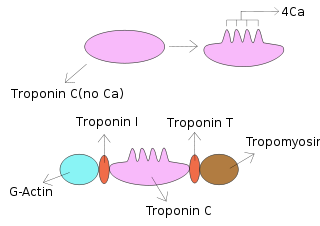
Troponin C is a protein which is part of the troponin complex. It contains four calcium-binding EF hands, although different isoforms may have fewer than four functional calcium-binding subdomains. It is a component of thin filaments, along with actin and tropomyosin. It contains an N lobe and a C lobe. The C lobe serves a structural purpose and binds to the N domain of troponin I (TnI). The C lobe can bind either Ca2+ or Mg2+. The N lobe, which binds only Ca2+, is the regulatory lobe and binds to the C domain of troponin I after calcium binding.

Troponin T is a part of the troponin complex, which are proteins integral to the contraction of skeletal and heart muscles. They are expressed in skeletal and cardiac myocytes. Troponin T binds to tropomyosin and helps position it on actin, and together with the rest of the troponin complex, modulates contraction of striated muscle. The cardiac subtype of troponin T is especially useful in the laboratory diagnosis of heart attack because it is released into the blood-stream when damage to heart muscle occurs. It was discovered by the German physician Hugo A. Katus at the University of Heidelberg, who also developed the troponin T assay.
Troponin I is a cardiac and skeletal muscle protein family. It is a part of the troponin protein complex, where it binds to actin in thin myofilaments to hold the actin-tropomyosin complex in place. Troponin I prevents myosin from binding to actin in relaxed muscle. When calcium binds to the troponin C, it causes conformational changes which lead to dislocation of troponin I. Afterwards, tropomyosin leaves the binding site for myosin on actin leading to contraction of muscle. The letter I is given due to its inhibitory character. It is a useful marker in the laboratory diagnosis of heart attack. It occurs in different plasma concentration but the same circumstances as troponin T - either test can be performed for confirmation of cardiac muscle damage and laboratories usually offer one test or the other.
An exonic splicing silencer (ESS) is a short region of an exon and is a cis-regulatory element. A set of 103 hexanucleotides known as FAS-hex3 has been shown to be abundant in ESS regions. ESSs inhibit or silence splicing of the pre-mRNA and contribute to constitutive and alternate splicing. To elicit the silencing affect, ESSs recruit proteins that will negatively affect the core splicing machinery.

Troponin I, cardiac muscle is a protein that in humans is encoded by the TNNI3 gene. It is a tissue-specific subtype of troponin I, which in turn is a part of the troponin complex.

Cardiac muscle troponin T (cTnT) is a protein that in humans is encoded by the TNNT2 gene. Cardiac TnT is the tropomyosin-binding subunit of the troponin complex, which is located on the thin filament of striated muscles and regulates muscle contraction in response to alterations in intracellular calcium ion concentration.

Tropomyosin alpha-1 chain is a protein that in humans is encoded by the TPM1 gene. This gene is a member of the tropomyosin (Tm) family of highly conserved, widely distributed actin-binding proteins involved in the contractile system of striated and smooth muscles and the cytoskeleton of non-muscle cells.

Tropomyosin alpha-3 chain is a protein that in humans is encoded by the TPM3 gene.

Troponin C, also known as TN-C or TnC, is a protein that resides in the troponin complex on actin thin filaments of striated muscle and is responsible for binding calcium to activate muscle contraction. Troponin C is encoded by the TNNC1 gene in humans for both cardiac and slow skeletal muscle.

β-Tropomyosin, also known as tropomyosin beta chain is a protein that in humans is encoded by the TPM2 gene. β-tropomyosin is striated muscle-specific coiled coil dimer that functions to stabilize actin filaments and regulate muscle contraction.
Troponin I, slow skeletal muscle is a protein that in humans is encoded by the TNNI1 gene. It is a tissue-specific subtype of troponin I, which in turn is a part of the troponin complex.

Troponin I, fast skeletal muscle is a protein that in humans is encoded by the TNNI2 gene.

Fast skeletal muscle troponin T (fTnT) is a protein that in humans is encoded by the TNNT3 gene.

Troponin C, skeletal muscle is a protein that in humans is encoded by the TNNC2 gene.

Enolase 3 (ENO3), more commonly known as beta-enolase (ENO-β), is an enzyme that in humans is encoded by the ENO3 gene.
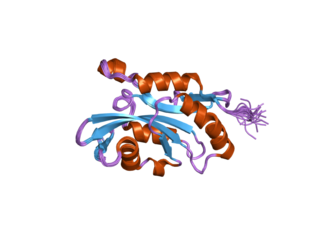
Cofilin 2 (muscle) also known as CFL2 is a protein which in humans is encoded by the CFL2 gene.







