
Actin is a family of globular multi-functional proteins that form microfilaments in the cytoskeleton, and the thin filaments in muscle fibrils. It is found in essentially all eukaryotic cells, where it may be present at a concentration of over 100 μM; its mass is roughly 42 kDa, with a diameter of 4 to 7 nm.
Troponin, or the troponin complex, is a complex of three regulatory proteins that are integral to muscle contraction in skeletal muscle and cardiac muscle, but not smooth muscle. Measurements of cardiac-specific troponins I and T are extensively used as diagnostic and prognostic indicators in the management of myocarditis, myocardial infarction and acute coronary syndrome. Blood troponin levels may be used as a diagnostic marker for stroke or other myocardial injury that is ongoing, although the sensitivity of this measurement is low.

Tropomyosin is a two-stranded alpha-helical, coiled coil protein found in many animal and fungal cells. In animals, it is an important component of the muscular system which works in conjunction with troponin to regulate muscle contraction. It is present in smooth and striated muscle tissues, which can be found in various organs and body systems, including the heart, blood vessels, respiratory system, and digestive system. In fungi, tropomyosin is found in cell walls and helps maintain the structural integrity of cells.
Nemaline myopathy is a congenital, often hereditary neuromuscular disorder with many symptoms that can occur such as muscle weakness, hypoventilation, swallowing dysfunction, and impaired speech ability. The severity of these symptoms varies and can change throughout one's life to some extent. The prevalence is estimated at 1 in 50,000 live births. It is the most common non-dystrophic myopathy.

MYH7 is a gene encoding a myosin heavy chain beta (MHC-β) isoform expressed primarily in the heart, but also in skeletal muscles. This isoform is distinct from the fast isoform of cardiac myosin heavy chain, MYH6, referred to as MHC-α. MHC-β is the major protein comprising the thick filament that forms the sarcomeres in cardiac muscle and plays a major role in cardiac muscle contraction.

Nebulin is an actin-binding protein which is localized to the thin filament of the sarcomeres in skeletal muscle. Nebulin in humans is coded for by the gene NEB. It is a very large protein and binds as many as 200 actin monomers. Because its length is proportional to thin filament length, it is believed that nebulin acts as a thin filament "ruler" and regulates thin filament length during sarcomere assembly. Other functions of nebulin, such as a role in cell signaling, remain uncertain.

Actin, alpha skeletal muscle is a protein that in humans is encoded by the ACTA1 gene.

Troponin I, cardiac muscle is a protein that in humans is encoded by the TNNI3 gene. It is a tissue-specific subtype of troponin I, which in turn is a part of the troponin complex.
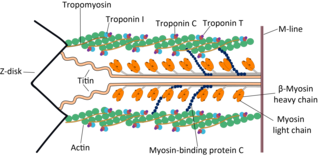
Cardiac muscle troponin T (cTnT) is a protein that in humans is encoded by the TNNT2 gene. Cardiac TnT is the tropomyosin-binding subunit of the troponin complex, which is located on the thin filament of striated muscles and regulates muscle contraction in response to alterations in intracellular calcium ion concentration.

Tropomyosin alpha-1 chain is a protein that in humans is encoded by the TPM1 gene. This gene is a member of the tropomyosin (Tm) family of highly conserved, widely distributed actin-binding proteins involved in the contractile system of striated and smooth muscles and the cytoskeleton of non-muscle cells.

Actin, cytoplasmic 2, or gamma-actin is a protein that in humans is encoded by the ACTG1 gene. Gamma-actin is widely expressed in cellular cytoskeletons of many tissues; in adult striated muscle cells, gamma-actin is localized to Z-discs and costamere structures, which are responsible for force transduction and transmission in muscle cells. Mutations in ACTG1 have been associated with nonsyndromic hearing loss and Baraitser-Winter syndrome, as well as susceptibility of adolescent patients to vincristine toxicity.

Tropomyosin alpha-3 chain is a protein that in humans is encoded by the TPM3 gene.

Troponin C, also known as TN-C or TnC, is a protein that resides in the troponin complex on actin thin filaments of striated muscle and is responsible for binding calcium to activate muscle contraction. Troponin C is encoded by the TNNC1 gene in humans for both cardiac and slow skeletal muscle.
Troponin I, slow skeletal muscle is a protein that in humans is encoded by the TNNI1 gene. It is a tissue-specific subtype of troponin I, which in turn is a part of the troponin complex.

Troponin I, fast skeletal muscle is a protein that in humans is encoded by the TNNI2 gene.

Slow skeletal muscle troponin T (sTnT) is a protein that in humans is encoded by the TNNT1 gene.

Myosin heavy chain, α isoform (MHC-α) is a protein that in humans is encoded by the MYH6 gene. This isoform is distinct from the ventricular/slow myosin heavy chain isoform, MYH7, referred to as MHC-β. MHC-α isoform is expressed predominantly in human cardiac atria, exhibiting only minor expression in human cardiac ventricles. It is the major protein comprising the cardiac muscle thick filament, and functions in cardiac muscle contraction. Mutations in MYH6 have been associated with late-onset hypertrophic cardiomyopathy, atrial septal defects and sick sinus syndrome.

Myosin essential light chain (ELC), ventricular/cardiac isoform is a protein that in humans is encoded by the MYL3 gene. This cardiac ventricular/slow skeletal ELC isoform is distinct from that expressed in fast skeletal muscle (MYL1) and cardiac atrial muscle (MYL4). Ventricular ELC is part of the myosin molecule and is important in modulating cardiac muscle contraction.

Troponin C, skeletal muscle is a protein that in humans is encoded by the TNNC2 gene.
Cofilin 2 (muscle) also known as CFL2 is a protein which in humans is encoded by the CFL2 gene.





