The 1,3-dipolar cycloaddition is a chemical reaction between a 1,3-dipole and a dipolarophile to form a five-membered ring. The earliest 1,3-dipolar cycloadditions were described in the late 19th century to the early 20th century, following the discovery of 1,3-dipoles. Mechanistic investigation and synthetic application were established in the 1960s, primarily through the work of Rolf Huisgen.[1][2] Hence, the reaction is sometimes referred to as the Huisgen cycloaddition (this term is often used to specifically describe the 1,3-dipolar cycloaddition between an organic azide and an alkyne to generate 1,2,3-triazole). 1,3-dipolar cycloaddition is an important route to the regio- and stereoselective synthesis of five-membered heterocycles and their ring-opened acyclic derivatives. The dipolarophile is typically an alkene or alkyne, but can be other pi systems. When the dipolarophile is an alkyne, aromatic rings are generally produced.
Originally two proposed mechanisms describe the 1,3-dipolar cycloaddition: first, the concerted pericycliccycloaddition mechanism, proposed by Rolf Huisgen;[3] and second, the stepwise mechanism involving a diradicalintermediate, proposed by Firestone.[4] After much debate, the former proposal is now generally accepted[5]—the 1,3-dipole reacts with the dipolarophile in a concerted, often asynchronous, and symmetry-allowed π4s + π2s fashion through a thermal six-electron Huckel aromatic transition state. However, a few examples exist of a stepwise mechanism for the catalyst-free 1,3-dipolar cycloaddition reactions of thiocarbonyl ylides,[6] and nitrile oxides[7]
The generic mechanism of a 1,3-dipolar cycloaddition between a dipole and a dipolarophile to give a five-membered heterocycle, through a six-electron transition state. Note that the red curly arrows are conventionally used to denote the reaction process but do not necessarily represent the actual flow of electrons.
Pericyclic mechanism
Huisgen investigated a series of cycloadditions between the 1,3-dipolar diazo compounds and various dipolarophilic alkenes.[3] The following observations support the concerted pericyclic mechanism, and refute the stepwise diradical or the stepwise polar pathway.
Substituent effects: Different substituents on the dipole do not exhibit a large effect on the cycloaddition rate, suggesting that the reaction does not involve a charge-separated intermediate.
Solvent effects: Solvent polarity has little effect on the cycloaddition rate, in line with the pericyclic mechanism where polarity does not change much in going from the reactants to the transition state.
Stereochemistry: 1,3-dipolar cycloadditions are always stereospecific with respect to the dipolarophile (i.e., cis-alkenes giving syn-products), supporting the concerted pericyclic mechanism in which two sigma bonds are formed simultaneously.
Structure and nomenclature of all second-row 1,3-dipoles consisting of carbon, nitrogen and oxygen centers. The dipoles are categorized as allyl-type or propargyl/allenyl-type based on the geometry of the central atom.
A 1,3-dipole is an organic molecule that can be represented as either an allyl-type or a propargyl/allenyl-type zwitterionic octet/sextet structures. Both types of 1,3-dipoles share four electrons in the π-system over three atoms. The allyl-type is bent whereas the propargyl/allenyl-type is linear in geometry.[8] 1,3-Dipoles containing higher-row elements such as sulfur or phosphorus are also known, but are utilized less routinely.
Resonance structures can be drawn to delocalize both negative and positive charges onto any terminus of a 1,3-dipole (see the scheme below). A more accurate method to describe the electronic distribution on a 1,3-dipole is to assign the major resonance contributor based on experimental or theoretical data, such as dipole moment measurements[9] or computations.[10] For example, diazomethane bears the largest negative character at the terminal nitrogen atom, while hydrazoic acid bears the largest negative character at the internal nitrogen atom.
Calculated major resonance structures of diazomethane and hydrazoic acid (doi = 10.1021/ja00475a007)
Consequently, this ambivalence means that the ends of a 1,3-dipole can be treated as both nucleophilic and electrophilic at the same time. The extent of nucleophilicity and electrophilicity at each end can be evaluated using the frontier molecular orbitals, which can be obtained computationally. In general, the atom that carries the largest orbital coefficient in the HOMO acts as the nucleophile, whereas that in the LUMO acts as the electrophile. The most nucleophilic atom is usually, but not always, the most electron-rich atom.[11][12][13] In 1,3-dipolar cycloadditions, identity of the dipole-dipolarophile pair determines whether the HOMO or the LUMO character of the 1,3-dipole will dominate (see discussion on frontier molecular orbitals below).
Dipolarophile
The most commonly used dipolarophiles are alkenes and alkynes. Heteroatom-containing dipolarophiles such as carbonyls and imines can also undergo 1,3-dipolar cycloaddition. Other examples of dipolarophiles include fullerenes and nanotubes, which can undergo 1,3-dipolar cycloaddition with azomethine ylide in the Prato reaction.
Solvent effects
1,3-Dipolar cycloadditions experience very little solvent effect because both the reactants and the transition states are generally non-polar. For example, the rate of reaction between phenyl diazomethane and ethyl acrylate or norbornene (see scheme below) changes only slightly upon varying solvents from cyclohexane to methanol.[14]
Effect of solvent polarity on 1,3-dipolar cycloaddition reactions(doi:10.3987/S(N)-1978-01-0109.)
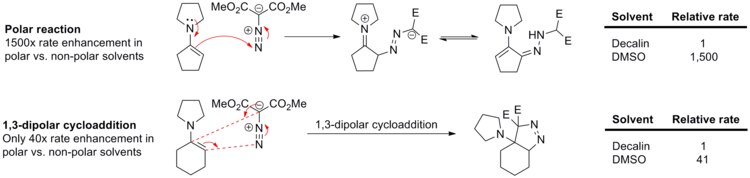
Lack of solvent effects in 1,3-dipolar cycloaddition is clearly demonstrated in the reaction between enamines and dimethyl diazomalonate (see scheme below).[15] The polar reaction, N-cyclopentenyl pyrrolidine nucleophilic addition to the diazo compound, proceeds 1,500 times faster in polar DMSO than in non-polar decalin. On the other hand, a close analog of this reaction, N-cyclohexenyl pyrrolidine 1,3-dipolar cycloaddition to dimethyl diazomalonate, is sped up only 41-fold in DMSO relative to decalin.
Rate of polar nucleophilic addition reaction versus 1,3-dipolar cycloaddition in decalin and in DMSO (doi:10.1016/S0040-4039(00)70991-9)
Frontier molecular orbital theory
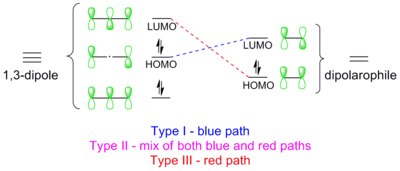
Orbital overlaps in types I, II and III 1,3-dipolar cycloaddition.
1,3-Dipolar cycloadditions are pericyclic reactions, which obey the Dewar-Zimmerman rules and the Woodward–Hoffmann rules. In the Dewar-Zimmerman treatment, the reaction proceeds through a 5-center, zero-node, 6-electron Huckel transition state for this particular molecular orbital diagram. However, each orbital can be randomly assigned a sign to arrive at the same result. In the Woodward–Hoffmann treatment, frontier molecular orbitals (FMO) of the 1,3-dipole and the dipolarophile overlap in the symmetry-allowed π4s + π2s manner. Such orbital overlap can be achieved in three ways: type I, II and III.[16] The dominant pathway is the one which possesses the smallest HOMO-LUMO energy gap.
Type I
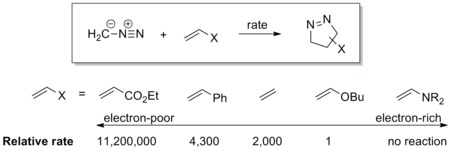
The dipole has a high-lying HOMO which overlaps with LUMO of the dipolarophile. A dipole of this class is referred to as a HOMO-controlled dipole or a nucleophilic dipole, which includes azomethine ylide, carbonyl ylide, nitrile ylide, azomethine imine, carbonyl imine and diazoalkane. These dipoles add to electrophilic alkenes readily. Electron-withdrawing groups (EWG) on the dipolarophile would accelerate the reaction by lowering the LUMO, while electron-donating groups (EDG) would decelerate the reaction by raising the HOMO. For example, the reactivity scale of diazomethane against a series of dipolarophiles is shown in the scheme below. Diazomethane reacts with the electron-poor ethyl acrylate more than a million times faster than the electron rich butyl vinyl ether.[17]
This type resembles the normal-electron-demand Diels-Alder reaction, in which the diene HOMO combines with the dienophile LUMO.
doi:10.1016/S0040-4039(01)92781-9
Type II
HOMO of the dipole can pair with LUMO of the dipolarophile; alternatively, HOMO of the dipolarophile can pair with LUMO of the dipole. This two-way interaction arises because the energy gap in either direction is similar. A dipole of this class is referred to as a HOMO-LUMO-controlled dipole or an ambiphilic dipole, which includes nitrile imide, nitrone, carbonyl oxide, nitrile oxide, and azide. Any substituent on the dipolarophile would accelerate the reaction by lowering the energy gap between the two interacting orbitals; i.e., an EWG would lower the LUMO while an EDG would raise the HOMO. For example, azides react with various electron-rich and electron-poor dipolarophile with similar reactivities (see reactivity scale below).[18]
doi:10.1021/ja01016a011
Type III
The dipole has a low-lying LUMO which overlaps with HOMO of the dipolarophile (indicated by red dashed lines in the diagram). A dipole of this class is referred to as a LUMO-controlled dipole or an electrophilic dipole, which includes nitrous oxide and ozone. EWGs on the dipolarophile decelerate the reaction, while EDGs accelerate the reaction. For example, ozone reacts with the electron-rich 2-methylpropene about 100,000 times faster than the electron-poor tetrachloroethene (see reactivity scale below).[19]
Concerted processes such as the 1,3-cycloaddition require a highly ordered transition state (high negative entropy of activation) and only moderate enthalpy requirements. Using competition reaction experiments, relative rates of addition for different cycloaddition reactions have been found to offer general findings on factors in reactivity.
Conjugation, especially with aromatic groups, increases the rate of reaction by stabilization of the transition state. During the transition, the two sigma bonds are being formed at different rates, which may generate partial charges in the transition state that can be stabilized by charge distribution into conjugated substituents.
More polarizable dipolarophiles are more reactive because diffuse electron clouds are better suited to initiate the flow of electrons.
Dipolarophiles with high angular strain are more reactive due to increased energy of the ground state.
Increased steric hindrance in the transition state as a result of unhindered reactants dramatically lowers the reaction rate.
Hetero-dipolarophiles add more slowly, if at all, compared to C,C-diapolarophiles due to a lower gain in sigma bond energy to offset the loss of a pi bond during the transition state.
Isomerism of the dipolarophile affects reaction rate due to sterics. trans-isomers are more reactive (trans-stilbene will add diphenyl(nitrile imide) 27 times faster than cis-stilbene) because during the reaction, the 120° bond angle shrinks to 109°, bringing eclipsing cis-substituents towards each other for increased steric clash.
1,3-dipolar cycloadditions usually result in retention of configuration with respect to both the 1,3-dipole and the dipolarophile. Such high degree of stereospecificity is a strong support for the concerted over the stepwise reaction mechanisms. As mentioned before, many examples show that the reactions were stepwise, thus, presenting partial or no stereospecificity.
With respect to dipolarophile
cis-Substituents on the dipolarophilic alkene end up cis, and trans-substituents end up trans in the resulting five-membered cyclic compound (see scheme below).[20]
doi:10.3987/S-1978-01-0147
With respect to dipole
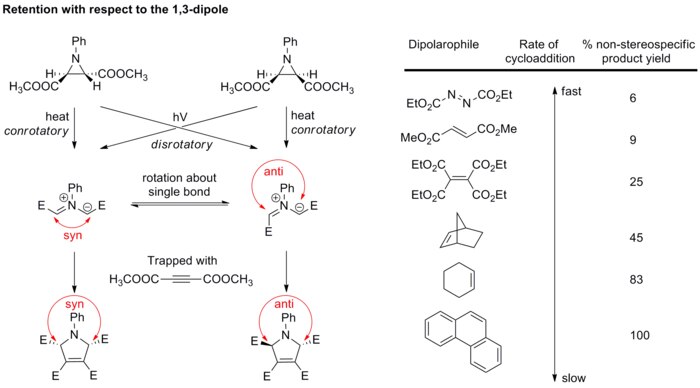
Generally, the stereochemistry of the dipole is not of major concern because only few dipoles could form stereogenic centers, and resonance structures allow bond rotation which scrambles the stereochemistry. However, the study of azomethine ylides has verified that cycloaddition is also stereospecific with respect to the dipole component. Diastereopure azomethine ylides are generated by electrocyclic ring opening of aziridines, and then rapidly trapped with strong dipolarophiles before bond rotation can take place (see scheme below).[21][22] If weaker dipolarophiles are used, bonds in the dipole have the chance to rotate, resulting in impaired cycloaddition stereospecificity.
These results altogether confirm that 1,3-dipolar cycloaddition is stereospecific, giving retention of both the 1,3-dipole and the dipolarophile.
doi:10.1021/ja00983a052
Diastereoselectivity
When two or more stereocenters are generated during the reaction, diastereomeric transition states and products can be obtained. In the Diels-Alder cycloaddition, the endodiastereoselectivity due to secondary orbital interactions is usually observed. In 1,3-dipolar cycloadditions, however, two forces influence the diastereoselectivity: the attractive π-interaction (resembling secondary orbital interactions in the Diels-Alder cycloaddition) and the repulsive steric interaction. Unfortunately, these two forces often cancel each other, causing poor diastereoselection in 1,3-dipolar cycloaddition.
Examples of substrate-controlled diastereoselective 1,3-dipolar cycloadditions are shown below. First is the reaction between benzonitrile N-benzylide and methyl acrylate. In the transition state, the phenyl and the methyl ester groups stack to give the cis-substitution as the exclusive final pyrroline product. This favorable π-interaction offsets the steric repulsion between the phenyl and the methyl ester groups.[23] Second is the reaction between nitrone and dihydrofuran. The exo-selectivity is achieved to minimize steric repulsion.[24] Last is the intramolecular azomethine ylide reaction with alkene. The diastereoselectivity is controlled by the formation of a less strained cis-fused ring system.[25]
doi:10.1021/ja00731a056
Directed 1,3-dipolar cycloaddition
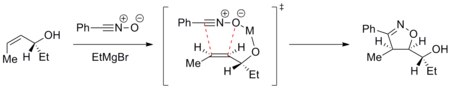
Trajectory of the cycloaddition can be controlled to achieve a diastereoselective reaction. For example, metals can chelate to the dipolarophile and the incoming dipole and direct the cycloaddition selectively on one face. The example below shows addition of nitrile oxide to an enantiomerically pure allyl alcohol in the presence of a magnesium ion. The most stable conformation of the alkene places the hydroxyl group above the plane of the alkene. The magnesium then chelates to the hydroxyl group and the oxygen atom of nitrile oxide. The cycloaddition thus comes from the top face selectively.[26]
Such diastereodirection has been applied in the synthesis of epothilones.[27]
The dominant electronic interaction is the combination between the largest HOMO and the largest LUMO. Therefore, regioselectivity is governed by the atoms that bear the largest orbital HOMO and LUMO coefficients.[29][30]
For example, consider the cycloaddition of diazomethane to three dipolarophiles: methyl acrylate, styrene or methyl cinnamate. The carbon of diazomethane bears the largest HOMO, while the end olefinic carbons of methyl acrylate and styrene bear the largest LUMO. Hence, cycloaddition gives the substitution at the C-3 position regioselectively. For methyl cinnamate, the two substituents (Ph v.s. COOMe) compete at withdrawing electrons from the alkene. The carboxyl is the better electron-withdrawing group, causing the β-carbon to be most electrophilic. Thus, cycloaddition yields the carboxyl group on C-3 and the phenyl group on C-4 regioselectively.
doi:10.1021/ja00444a013 and doi:10.1021/ja00436a062
Steric effect
Steric effects can either cooperate or compete with the aforementioned electronic effects. Sometimes steric effects completely outweighs the electronic preference, giving the opposite regioisomer exclusively.[31]
For example, diazomethane generally adds to methyl acrylate to give 3-carboxyl pyrazoline. However, by putting more steric demands into the system, we start to observe the isomeric 4-carboxyl pyrazolines. The ratio of these two regioisomers depends on the steric demands. At the extreme, increasing the size from hydrogen to t-butyl shifts the regioselectivity from 100% 3-carboxyl to 100% 4-carboxyl substitution.[32][33]
1,3-dipolar cycloadditions are important ways toward the synthesis of many important 5-membered heterocycles such as triazoles, furans, isoxazoles, pyrrolidines, and others. Additionally, some cycloadducts can be cleaved to reveal the linear skeleton, providing another route toward the synthesis of aliphatic compounds. These reactions are tremendously useful also because they are stereospecific, diastereoselective and regioselective. Several examples are provided below.
Nitrile oxides
1,3-dipolar cycloaddition with nitrile oxides is a widely used masked-aldol reaction. Cycloaddition between a nitrile oxide and an alkene yields the cyclic isoxazoline product, whereas the reaction with an alkyne yields the isoxazole. Both isoxazolines and isoxazoles can be cleaved by hydrogenation to reveal aldol-type β-hydroxycarbonyl or Claisen-type β-dicarbonyl products, respectively.
Nitrile oxide-alkyne cycloaddition followed by hydrogenation was utilized in the synthesis of Miyakolide as illustrated in the figure below.[34]
Carbonyl ylides
1,3-dipolar cycloaddition reactions have emerged as powerful tools in the synthesis of complex cyclic scaffolds and molecules for medicinal, biological, and mechanistic studies. Among them, [3+2] cycloaddition reactions involving carbonyl ylides have extensively been employed to generate oxygen-containing five-membered cyclic molecules.[35]
Preparation of carbonyl ylides for 1,3-dipolar cycloaddition reactions
Ylides are regarded as positively charged heteroatoms connected to negatively charged carbon atoms, which include ylides of sulfonium, thiocarbonyl, oxonium, nitrogen, and carbonyl.[36] Several methods exist for generating carbonyl ylides, which are necessary intermediates for generating oxygen-containing five-membered ring structures, for [3+2] cycloaddition reactions.
Synthesis of carbonyl ylides from diazomethane derivatives by photocatalysis
Scheme 1. Photolysis of DTTC in the presence of tetramethylurea. Modified from Janulis, E. P.; Arduengo, A. J. J. Am. Chem. Soc. 1983, 105, 5929.
Another early example of carbonyl ylide synthesis by photocatalysis was reported by Olah et al.[38] Dideuteriodiazomethane was photolysed in the presence of formaldehyde to generate the dideuterioformaldehyde carbonyl ylide.
Scheme 2. Photolysis of dideuteriodiazomethane with formaldehyde. Modified from Prakash, G. K. S.; Ellis, R. W.; Felberg, J. D.; Olah, G. A. J Am Chem Soc 1986, 108, 1341.
Synthesis of carbonyl ylides from hydroxypyrones by proton transfer
Carbonyl ylides can be synthesized by acid catalysis of hydroxy-3-pyrones in the absence of a metal catalyst.[39] An initial tautomerization occurs, followed by elimination of the leaving group to aromatize the pyrone ring and to generate the carbonyl ylide. A cycloaddition reaction with a dipolarophile lastly forms the oxacycle. This approach is less widely employed due to its limited utility and requirement for pyrone skeletons.
Scheme 3. Acid-catalyzed synthesis of carbonyl ylides from hydroxy-3-pyrones. Modified from Sammes, P. G.; Street, L. J. J. Chem. Soc., Chem. Commun. 1982, 1056.
5-hydroxy-4-pyrones can also be used to synthesize carbonyl ylides by an intramolecularhydrogen transfer.[40] After hydrogen transfer, the carbonyl ylide can then react with dipolarophiles to form oxygen-containing rings.
Scheme 4. Intramolecular hydrogen transfer-mediated synthesis of carbonyl ylides from 5-hydroxy-4-pyrones. Modified from Garst, M. E.; McBride, B. J.; Douglass III, J. G. Tetrahedron Lett. 1983, 24, 1675.
Synthesis of α-halocarbonyl ylides from dihalocarbenes
Dihalocarbenes have also been employed to generate carbonyl ylides, exploiting the electron withdrawing nature of dihalocarbenes.[41][42][43] Both phenyl(trichloromethyl)mercury and phenyl(tribromomethyl)mercury are sources dichlorocarbenes and dibromocarbenes, respectively. The carbonyl ylide can be generated upon reaction of the dihalocarbenes with ketones or aldehydes. However, the synthesis of α-halocarbonyl ylides can also undesirably lead to the loss of carbon monoxide and the generation of the deoxygenation product.
Scheme 5. α-Halocarbonyl ylide synthesis through dihalocarbene intermediates. Modified from Padwa, A.; Hornbuckle, S. F. Chem Rev 1991, 91, 263.
Synthesis of carbonyl ylides from diazomethane derivatives by metal catalysis
A universal approach for generating carbonyl ylides involves metal catalysis of α-diazocarbonyl compounds, generally in the presence of dicopper or dirhodium catalysts.[44] After release of nitrogen gas and conversion to the metallocarbene, an intermolecular reaction with a carbonyl group can generate the carbonyl ylide. Subsequent cycloaddition reaction with an alkene or alkyne dipolarophile can afford oxygen-containing five-membered rings. Popular catalysts that give modest yields towards synthesizing oxacycles include Rh2(OAc)4 and Cu(acac)2.[45][46]
Scheme 6. Metal-catalyzed synthesis of carbonyl ylides. Reproduced from Hodgson, D. M.; Bruckl, T.; Glen, R.; Labande, A. H.; Selden, D. A.; Dossetter, A. G.; Redgrave, A. J. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 5450.
Mechanism of the 1,3-dipolar cycloaddition reaction mediated by metal catalysis of diazocarbonyl compounds
The universality and extensive use of 1,3-dipolar cycloaddition reactions mediated by metal catalysis of diazocarbonyl molecules, for synthesizing oxygen-containing five-membered rings, has spurred significant interest into its mechanism. Several groups have investigated the mechanism to expand the scope of synthetic molecules with respect to regio- and stereo-selectivity. However, due to the high turn over frequencies of these reactions, the intermediates and mechanism remains elusive. The generally accepted mechanism, developed by characterization of stable ruthenium-carbenoid complexes[47] and rhodium metallocarbenes,[48] involves an initial formation of a metal-carbenoid complex from the diazo compound. Elimination of nitrogen gas then affords a metallocarbene. An intramolecular nucleophilic attack by the carbonyl oxygen regenerates the metal catalyst and forms the carbonyl ylide. The carbonyl ylide can then react with an alkene or alkyne, such as dimethyl acetylenedicarboxylate (DMAD) to generate the oxacycle.
Scheme 7. Accepted mechanism of the 1,3-dipolar cycloaddition reaction mediated by metal catalysis (example dirhodium catalyst) of diazocarbonyl compounds. Modified from M. Hodgson, D.; H. Labande, A.; Muthusamy, S. In Organic Reactions; John Wiley & Sons, Inc.: 2004.
However, it is uncertain whether the metallocarbene intermediate generates the carbonyl ylide. In some cases, metallocarbenes can also react directly with dipolarophiles.[49] In these cases, the metallocarbene, such as the dirhodium(II)tetracarboxylate carbene, is stabilized through hyperconjugative metal enolate-type interactions.[50][51][52][53] Subsequent 1,3-dipolar cycloaddition reaction occurs through a transient metal-complexed carbonyl ylide. Therefore, a persistent metallocarbene can influence the stereoselectivity and regioselectivity of the 1,3-dipolar cycloaddition reaction based on the stereochemistry and size of the metal ligands.
The dirhodium(II)tetracarboxylate metallocarbene stabilized by πC-Rh→πC=O hyperconjugation. Modified from M. Hodgson, D.; H. Labande, A.; Muthusamy, S. In Organic Reactions; John Wiley & Sons, Inc.: 2004.
Scheme 9. Products of the 1,3-dipolar cycloaddition reaction between carbonyl ylide dipoles and alkenyl or alkynyl dipolarophiles. Modified from M. Hodgson, D.; H. Labande, A.; Muthusamy, S. In Organic Reactions; John Wiley & Sons, Inc.: 2004.
Regioselectivity of the 1,3-dipolar cycloaddition reaction mediated by metal catalysis of diazocarbonyl compounds
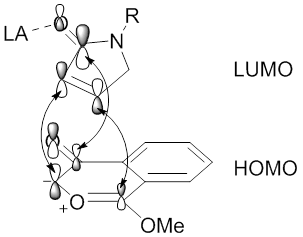
Regioselectivity of 1,3-dipolar cycloaddition reactions between carbonyl ylide dipoles and alkynyl or alkenyl dipolarophiles is essential for generating molecules with defined regiochemistry. FMO theory and analysis of the HOMO-LUMO energy gaps between the dipole and dipolarophile can rationalize and predict the regioselectivity of experimental outcomes.[56][57] The HOMOs and LUMOs can belong to either the dipole or dipolarophile, for which HOMOdipole-LUMOdipolarophile or HOMOdipolarophile-LUMOdipole interactions can exist. Overlap of the orbitals with the largest coefficients can ultimately rationalize and predict results.
Scheme 10. diagram of the molecular orbital interactions of HOMOdipole-LUMOdipolarophile or HOMOdipolarophile-LUMOdipole between a carbonyl ylide dipole and alkenyl dipolarophile.
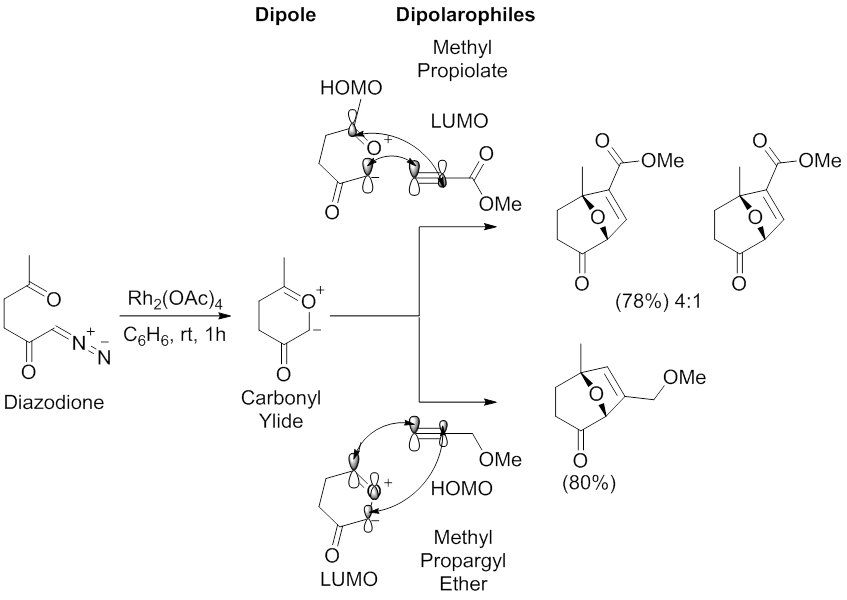
The archetypal regioselectivity of the 1,3-dipolar cycloaddition reaction mediated by carbonyl ylide dipoles has been examined by Padwa and coworkers.[55][58] Using a Rh2(OAc)4 catalyst in benzene, diazodione underwent a 1,3-dipolar cycloaddition reaction with methyl propiolate and methyl propargylether. The reaction with methyl propiolate affords two regioisomers with the major resulting from the HOMOdipole-LUMOdipolarophile interaction, which has the largest coefficients on the carbon proximal to the carbonyl group of the carbonyl ylide and on the methyl propiolate terminal alkyne carbon. The reaction with methyl propargyl ether affords one regioisomer resulting from the HOMOdipolarophile-LUMOdipole interaction, which has largest coefficients on the carbon distal to the carbonyl group of the carbonyl ylide and on the methyl propargyl ether terminal alkyne carbon.
Scheme 11. Regioselectivity and molecular orbital interactions of the 1,3-dipolar cycloaddition reaction between a diazodione and methyl propiolate or methyl propargyl ether. Modified from Padwa, A.; Weingarten, M. D. Chem Rev 1996, 96, 223.
Regioselectivities of 1,3-dipolar cycloaddition reactions mediated by metal catalysis of diazocarbonyl compounds may also be influenced by the metal through formation of stable metallocarbenes.[49][59] Stabilization of the metallocarbene, via metal enolate-type interactions, will prevent the formation of carbonyl ylides, resulting in a direct reaction between the metallocarbene dipole and an alkynyl or alkenyl dipolarophile (see image of The dirhodium(II)tetracarboxylate metallocarbene stabilized by πC-Rh→πC=O hyperconjugation.). In this situation, the metal ligands will influence the regioselectivity and stereoselectivity of the 1,3-dipolar cycloaddition reaction.
Stereoselectivity and asymmetric induction of the 1,3-dipolar cycloaddition reaction mediated by metal catalysis of diazocarbonyl compounds
The stereoselectivity of 1,3-dipolar cycloaddition reactions between carbonyl ylide dipoles and alkenyl dipolarophiles has also been closely examined. For alkynyl dipolarophiles, stereoselectivity is not an issue as relatively planar sp2 carbons are formed, while regioselectivity must be considered (see image of the Products of the 1,3-Dipolar Cycloaddition Reaction Between Carbonyl Ylide Dipoles and Alkenyl or Alkynyl Dipolarophiles). However, for alkenyl dipolarophiles, both regioselectivity and stereoselectivity must be considered as sp3 carbons are generated in the product species.
1,3-dipolar cycloaddition reactions between carbonyl ylide dipoles and alkenyl dipolarophiles can generate diastereomeric products.[53] The exo product is characterized with dipolarophile substituents being cis to the ether bridge of the oxacycle. The endo product is characterized with the dipolarophile substituents being trans to the ether bridge of the oxacycle. Both products can be generated through pericyclic transitions states involving concerted synchronous or concerted asynchronous processes.
One early example conferred stereoselectivity in terms of endo and exo products with metal catalysts and Lewis acids.[60] Reactions with just the metal catalyst Rh2(OAc)4 prefer the exo product while reactions with the additional Lewis acid Yb(OTf)3 prefer the endo product. The endo selectivity observed for Lewis acid cycloaddition reactions is attributed to the optimized orbital overlap of the carbonyl π systems between the dipolarophile coordinated by Yb(Otf)3 (LUMO) and the dipole (HOMO). After many investigations, two primary approaches for influencing the stereoselectivity of carbonyl ylide cycloadditions have been developed that exploit the chirality of metal catalysts and Lewis acids.[53]
Facial Selectivity of the 1,3-Dipolar Cycloaddition Reaction using a Metal Catalyst and Lewis AcidRationale for the Endo Selectivity of the 1,3-Dipolar Cycloaddition Reaction with a Lewis Acid
The first approach employs chiral metal catalysts to modulate the endo and exo stereoselectivity. The chiral catalysts, in particular Rh2[(S)-DOSP]4 and Rh2[(S)-BPTV]4 can induce modest asymmetric induction and was used to synthesize the antifungal agent pseudolaric acid A.[61] This is a result of the chiral metal catalyst remaining associated with the carbonyl ylide during the cycloaddition, which confers facial selectivity. However, the exact mechanisms are not yet fully understood.
Asymmetric induction of the 1,3-dipolar cycloaddition reaction with chiral metal catalysts
The second approach employs a chiral Lewis acid catalyst to induce facial stereoselectivity after the generation of the carbonyl ylide using an achiral metal catalyst.[62] The chiral Lewis acid catalyst is believed to coordinate to the dipolarophile, which lowers the LUMO of the dipolarophile while also leading to enantioselectivity.
Asymmetric induction of the 1,3-dipolar cycloaddition reaction with chiral Lewis acid catalysts
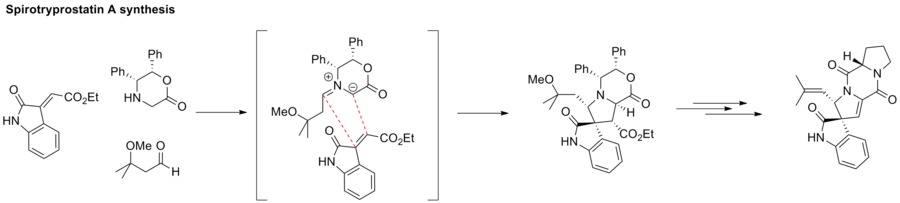
1,3-Dipolar cycloaddition between an azomethine ylide and an alkene furnishes an azacyclic structure, such as pyrrolidine. This strategy has been applied to the synthesis of spirotryprostatin A.[63]
The 1,3-dipolar cycloaddition between organic azides and terminal alkynes (i.e., the Huisgen cycloaddition) has been widely utilized for bioconjugation.
Copper catalysis
The Huisgen reaction generally does not proceed readily under mild conditions. Meldal et al. and Sharpless et al. independently developed a copper(I)-catalyzed version of the Huisgen reaction, CuAAC (for Copper-catalyzed Azide-Alkyne Cycloaddition), which proceeds very readily in mild, including physiological, conditions (neutral pH, room temperature and water solution).[64][65] This reaction is also bioorthogonal: azides and alkynes are both generally absent from biological systems and therefore these functionalities can be chemoselectively reacted even in the cellular context. They also do not react with other functional groups found in nature, so they do not perturb biological systems. The reaction is so versatile that it is termed the "Click" chemistry. Although copper(I) is toxic, many protective ligands have been developed to both reduce cytotoxicity and improve rate of CuAAC, allowing it to be used in in vivo studies.[66]
To avoid toxicity of copper(I), Bertozzi et al. developed the strain-promoted azide-alkyne cycloaddition (SPAAC) between organic azide and strained cyclooctyne. The angle distortion of the cyclooctyne helps to speed up the reaction by both reducing the activation strain and enhancing the interactions, thereby enabling it to be used in physiological conditions without the need for the catalyst.[68]
For instance, Ting et al. introduced an azido functionality onto specific proteins on the cell surface using a ligase enzyme. The azide-tagged protein is then labeled with cyclooctyne-fluorophore conjugate to yield a fluorescently labeled protein.[69]
References
↑ Bertrand, Guy; Wentrup, Curt (17 March 1994). "Nitrile Imines: From Matrix Characterization to Stable Compounds". Angewandte Chemie International Edition in English. 33 (5): 527–545. doi:10.1002/anie.199405271.
↑ Huisgen, Rolf (October 1963). "1,3-Dipolar Cycloadditions. Past and Future". Angewandte Chemie International Edition in English. 2 (10): 565–598. doi:10.1002/anie.196305651.
↑ Seyyed Amir, Siadati (2015). "An example of a stepwise mechanism for the catalyst-free 1,3-dipolar cycloaddition between a nitrile oxide and an electron rich alkene". Tetrahedron Letters. 56 (34): 4857–4863. doi:10.1016/j.tetlet.2015.06.048.
↑ Williamson, D. G.; Cvetanovic, R. J. (1968). "Rates of ozone-olefin reactions in carbon tetrachloride solutions". Journal of the American Chemical Society. 90 (14): 3668–3672. Bibcode:1968JAChS..90.3668W. doi:10.1021/ja01016a011.
↑ Dahmen, Alexander; Hamberger, Helmut; Huisgen, Rolf; Markowski, Volker (1971). "Conrotatory ring opening of cyanostilbene oxides to carbonyl ylides". Journal of the Chemical Society D: Chemical Communications (19): 1192–1194. doi:10.1039/C29710001192.
↑ Padwa, Albert; Smolanoff, Joel (1971). "Photocycloaddition of arylazirenes with electron-deficient olefins". Journal of the American Chemical Society. 93 (2): 548–550. Bibcode:1971JAChS..93..548P. doi:10.1021/ja00731a056.
↑ Wang, Chia-Lin; Ripka, William; Confalone, Pat (1984). "A short and stereospecific synthesis of (±)-α-lycorane". Tetrahedron Letters. 25 (41): 4613–4616. doi:10.1016/S0040-4039(01)91213-4.
↑ Bode, Jeffrey; Carreira, Erick (2011). "Stereoselective Syntheses of Epothilones A and B via Directed Nitrile Oxide Cycloaddition". Journal of the American Chemical Society. 123 (15): 3611–3612. doi:10.1021/ja0155635. PMID11472140.
↑ Caramella, Pierluigi; Houk, K.N. (1976). "Geometries of nitrilium betaines. The clarification of apparently anomalous reactions of 1,3-dipoles". Journal of the American Chemical Society. 98 (20): 6397–6399. Bibcode:1976JAChS..98.6397C. doi:10.1021/ja00436a062.
↑ Caramella, Pierluigi; Gandour, Ruth W.; Hall, Janet A.; Deville, Cynthia G.; Houk, K. N. (1977). "A derivation of the shapes and energies of the molecular orbitals of 1,3-dipoles. Geometry optimizations of these species by MINDO/2 and MINDO/3". Journal of the American Chemical Society. 99 (2): 385–392. Bibcode:1977JAChS..99..385C. doi:10.1021/ja00444a013.
↑ Padwa, Albert (1983). 1,3-Dipolar Cycloaddition Chemistry. General Heterocyclic Chemistry Series. Vol.1. United States of America: Wiley-Interscience. pp.141–145. ISBN978-0-471-08364-1.
↑ Synthetic Reactions of M=C and M=N Bonds: Ylide Formation, Rearrangement, and 1,3-Dipolar Cycloaddition; Hiyama, T. W., J., Ed.; Elsevier, 2007; Vol. 11.
↑ Padwa, Albert.; Hornbuckle, Susan F. (1991). "Ylide formation from the reaction of carbenes and carbenoids with heteroatom lone pairs". Chemical Reviews. 91 (3): 263–309. doi:10.1021/cr00003a001.
1 2 Janulis, Eugene P.; Arduengo, Anthony J. (1983). "Structure of an electronically stabilized carbonyl ylide". Journal of the American Chemical Society. 105 (18): 5929–5930. Bibcode:1983JAChS.105.5929J. doi:10.1021/ja00356a044.
↑ Prakash, G. K. S.; Ellis, R. W.; Felberg, J. D.; Olah, G. A. Formaldehyde 0-Methylide, [CH2=O+-CH2: The Parent Carbonyl Ylide] J Am Chem Soc 1986, 108, 1341.
↑ Huan, Zhenwei; Landgrebe, John A.; Peterson, Kimberly (1983). "Dibromocarbonyl ylides. Deoxygenation of aldehydes and ketones by dibromocarbene". The Journal of Organic Chemistry. 48 (24): 4519–4523. doi:10.1021/jo00172a015.
↑ Martin, Charles W.; Lund, Paul R.; Rapp, Erich; Landgrebe, John A. (1978). "Halogenated carbonyl ylides in the reactions of mercurial dihalocarbene precursors with substituted benzaldehydes". The Journal of Organic Chemistry. 43 (6): 1071–1076. doi:10.1021/jo00400a009.
↑ Padwa, Albert; Hertzog, Donald L.; Nadler, William R. (1994). "Intramolecular Cycloaddition of Isomunchnone Dipoles to Heteroaromatic .pi.-Systems". The Journal of Organic Chemistry. 59 (23): 7072–7084. doi:10.1021/jo00102a037.
↑ Park, Soon-Bong; Sakata, Naoya; Nishiyama, Hisao (1996). "Aryloxycarbonylcarbene Complexes of Bis(oxazolinyl)pyridineruthenium as Active Intermediates in Asymmetric Catalytic Cyclopropanations". Chemistry - A European Journal. 2 (3): 303–306. doi:10.1002/chem.19960020311.
↑ Snyder, James P.; Padwa, Albert; Stengel, Thomas; Arduengo, Anthony J.; Jockisch, Alexander; Kim, Hyo-Joong (2001). "A Stable Dirhodium Tetracarboxylate Carbenoid: Crystal Structure, Bonding Analysis, and Catalysis". Journal of the American Chemical Society. 123 (45): 11318–11319. Bibcode:2001JAChS.12311318S. doi:10.1021/ja016928o. PMID11697986.
↑ Yoshikai, Naohiko; Nakamura, Eiichi (2003). "Theoretical Studies on Diastereo- and Enantioselective Rhodium-Catalyzed Cyclization of Diazo Compoundvia Intramolecular C—H Bond Insertion". Advanced Synthesis & Catalysis. 345 (910): 1159–1171. doi:10.1002/adsc.200303092.
↑ Nakamura, Eiichi; Yoshikai, Naohiko; Yamanaka, Masahiro (2002). "Mechanism of C−H Bond Activation/C−C Bond Formation Reaction between Diazo Compound and Alkane Catalyzed by Dirhodium Tetracarboxylate". Journal of the American Chemical Society. 124 (24): 7181–7192. Bibcode:2002JAChS.124.7181N. doi:10.1021/ja017823o. PMID12059244.
1 2 3 4 M. Hodgson, D.; H. Labande, A.; Muthusamy, S. In Organic Reactions; John Wiley & Sons, Inc.: 2004.
↑ Suga, Hiroyuki; Ebiura, Yasutaka; Fukushima, Kazuaki; Kakehi, Akikazu; Baba, Toshihide (2005). "Efficient Catalytic Effects of Lewis Acids in the 1,3-Dipolar Cycloaddition Reactions of Carbonyl Ylides with Imines". The Journal of Organic Chemistry. 70 (26): 10782–10791. doi:10.1021/jo051743b. PMID16356001.
1 2 Padwa, Albert; Fryxell, Glen E.; Zhi, Lin (1990). "Tandem cyclization-cycloaddition reaction of rhodium carbenoids. Scope and mechanistic details of the process". Journal of the American Chemical Society. 112 (8): 3100–3109. Bibcode:1990JAChS.112.3100P. doi:10.1021/ja00164a034.
↑ Houk, K. N.; Sims, Joyner.; Duke, R. E.; Strozier, R. W.; George, John K. (1973). "Frontier molecular orbitals of 1,3 dipoles and dipolarophiles". Journal of the American Chemical Society. 95 (22): 7287–7301. Bibcode:1973JAChS..95.7287H. doi:10.1021/ja00803a017.
↑ Houk, K. N.; Rondan, Nelson G.; Santiago, Cielo; Gallo, Catherine J.; Gandour, Ruth Wells; Griffin, Gary W. (1980). "Theoretical studies of the structures and reactions of substituted carbonyl ylides". Journal of the American Chemical Society. 102 (5): 1504–1512. Bibcode:1980JAChS.102.1504H. doi:10.1021/ja00525a006.
↑ Padwa, Albert; Weingarten, M. David (1996). "Cascade Processes of Metallo Carbenoids". Chemical Reviews. 96 (1): 223–270. doi:10.1021/cr950022h. PMID11848752.
↑ Padwa, Albert; Austin, David J. (1996). "Ligand-Induced Selectivity in the Rhodium(II)-Catalyzed Reactions of α-Diazo Carbonyl Compounds†". The Journal of Organic Chemistry. 61: 63–72. doi:10.1021/jo951576n.
↑ Agard, Nicholas; Prescher, Jennifer; Bertozzi, Carolyn (2004). "A Strain-Promoted [3 + 2] Azide−Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems". Journal of the American Chemical Society. 126 (46): 15046–15047. Bibcode:2004JAChS.12615046A. doi:10.1021/ja044996f. PMID15547999.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.