 | |
| Clinical data | |
|---|---|
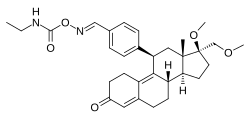
| Other names | J-956; 11β-(4-((E)-(Ethylcarbamoyl-oxyimino)methyl)phenyl)-17β-methoxy-17α-(methoxymethyl)estra-4,9-dien-3-one |
| Identifiers | |
| |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C31H40N2O5 |
| Molar mass | 520.670 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
Asoprisnil ecamate (INN; development code J-956) is a synthetic, steroidal selective progesterone receptor modulator (SPRM) which was under development for the treatment of endometriosis, uterine fibroids, and menopausal symptoms but was discontinued. [1] [2] [3] It is a potent and highly selective ligand of the progesterone receptor with mixed agonistic and antagonistic activity and much reduced antiglucocorticoid activity relative to mifepristone. [2] [3] [4] The drug reached phase III clinical trials for the aforementioned indications prior to its discontinuation. [1]