Electrophilic fluorination is the combination of a carbon-centered nucleophile with an electrophilic source of fluorine to afford organofluorine compounds. Although elemental fluorine and reagents incorporating an oxygen-fluorine bond can be used for this purpose, they have largely been replaced by reagents containing a nitrogen-fluorine bond. [1]
Contents
- Mechanism and stereochemistry
- Prevailing mechanism
- Stereoselective variants
- Scope and limitations
- Fluorinating reagents
- Nucleophilic substrates
- Comparison with other methods
- Typical conditions
- See also
- References
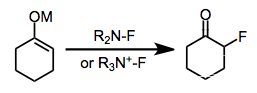
Electrophilic fluorination offers an alternative to nucleophilic fluorination methods employing alkali or ammonium fluorides and methods employing sulfur fluorides for the preparation of organofluorine compounds. Development of electrophilic fluorination reagents has always focused on removing electron density from the atom attached to fluorine; however, compounds containing nitrogen-fluorine bonds have proven to be the most economical, stable, and safe electrophilic fluorinating agents. Electrophilic N-F reagents are either neutral or cationic and may possess either sp2- or sp3-hybridized nitrogen. Although the precise mechanism of electrophilic fluorination is currently unclear, highly efficient and stereoselective methods have been developed.
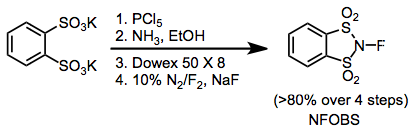
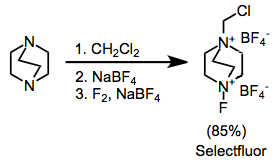
Some common fluorinating agents used for organic synthesis are N-fluoro-o-benzenedisulfonimide (NFOBS), N-fluorobenzenesulfonimide (NFSI), and Selectfluor. [1]