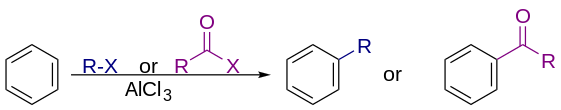In commercial applications, the alkylating agents are generally alkenes, some of the largest scale reactions practiced in industry. Such alkylations are of major industrial importance, e.g. for the production of ethylbenzene, the precursor to polystyrene, from benzene and ethylene and for the production of cumene from benzene and propene in cumene process:
Industrial production typically uses solid acids derived from a zeolite as the catalyst.
With alkyl halides
Friedel–Crafts alkylation involves the alkylation of an aromatic ring. Traditionally, the alkylating agents are alkyl halides. Many alkylating agents can be used instead of alkyl halides. For example, enones and epoxides can be used in presence of protons. The reaction typically employs a strong Lewis acid, such as aluminium chloride as catalyst, to increase the electrophilicity of the alkylating agent.[6]
This reaction suffers from the disadvantage that the product is more nucleophilic than the reactant because alkyl groups are activators for the Friedel–Crafts reaction. Consequently, overalkylation can occur. However, steric hindrance can be exploited to limit the number of successive alkylation cycles that occur, as in the t-butylation of 1,4-dimethoxybenzene that gives only the product of two alkylation cycles and with only one of three possible isomers of it:[7]
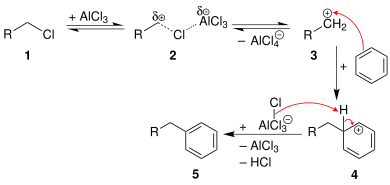
Furthermore, the reaction is only useful for primary alkyl halides in an intramolecular sense when a 5- or 6-membered ring is formed. For the intermolecular case, the reaction is limited to tertiary alkylating agents, some secondary alkylating agents (ones for which carbocation rearrangement is degenerate), or alkylating agents that yield stabilized carbocations (e.g., benzylic or allylic ones). In the case of primary alkyl halides, the carbocation-like complex (R(+)---X---Al(-)Cl3) will undergo a carbocationrearrangement reaction to give almost exclusively the rearranged product derived from a secondary or tertiary carbocation.[8]
The general mechanism for primary alkyl halides is shown in the figure below.[8]
Mechanism of Friedel–Crafts alkylation.For primary (and possibly secondary) alkyl halides, a carbocation-like complex with the Lewis acid, [R(+)---(X---MXn)(–)] is more likely to be involved, rather than a free carbocation.
Friedel–Crafts dealkylation
Friedel–Crafts alkylations can be reversible. Although this is usually undesirable it can be exploited; for instance by facilitating transalkylation reactions.[10]
1,3-Diisopropylbenzene is produced via transalkylation, a special form of Friedel–Crafts alkylation.
Friedel–Crafts acylation involves the acylation of aromatic rings. Typical acylating agents are acyl chlorides. Acid anhydrides as well as carboxylic acids are also viable. A typical Lewis acid catalyst is aluminium trichloride. Because, however, the product ketone forms a rather stable complex with Lewis acids such as AlCl3, a stoichiometric amount or more of the "catalyst" must generally be employed, unlike the case of the Friedel–Crafts alkylation, in which the catalyst is constantly regenerated.[13] Reaction conditions are similar to the Friedel–Crafts alkylation. This reaction has several advantages over the alkylation reaction. Due to the electron-withdrawing effect of the carbonyl group, the ketone product is always less reactive than the original molecule, so multiple acylations do not occur. Also, there are no carbocation rearrangements, as the acylium ion is stabilized by a resonance structure in which the positive charge is on the oxygen.
The viability of the Friedel–Crafts acylation depends on the stability of the acyl chloride reagent. Formyl chloride, for example, is too unstable to be isolated. Thus, synthesis of benzaldehyde through the Friedel–Crafts pathway requires that formyl chloride be synthesized in situ. This is accomplished by the Gattermann-Koch reaction, accomplished by treating benzene with carbon monoxide and hydrogen chloride under high pressure, catalyzed by a mixture of aluminium chloride and cuprous chloride. Simple ketones that could be obtained by Friedel–Crafts acylation are produced by alternative methods, e.g., oxidation, in industry.
Reaction mechanism
The reaction proceeds through generation of an acylium center. The reaction is completed by deprotonation of the arenium ion by AlCl4−, regenerating the AlCl3 catalyst. However, in contrast to the truly catalytic alkylation reaction, the formed ketone is a moderate Lewis base, which forms a complex with the strong Lewis acid aluminum trichloride. The formation of this complex is typically irreversible under reaction conditions. Thus, a stochiometric quantity of AlCl3 is needed. The complex is destroyed upon aqueous workup to give the desired ketone. For example, the classical synthesis of deoxybenzoin calls for 1.1 equivalents of AlCl3 with respect to the limiting reagent, phenylacetyl chloride.[14] In certain cases, generally when the benzene ring is activated, Friedel–Crafts acylation can also be carried out with catalytic amounts of a milder Lewis acid (e.g. Zn(II) salts) or a Brønsted acid catalyst using the anhydride or even the carboxylic acid itself as the acylation agent.
If desired, the resulting ketone can be subsequently reduced to the corresponding alkane substituent by either Wolff–Kishner reduction or Clemmensen reduction. The net result is the same as the Friedel–Crafts alkylation except that rearrangement is not possible.[15]
Hydroxyalkylation
Arenes react with certain aldehydes and ketones to form the hydroxyalkylated products, for example in the reaction of the mesityl derivative of glyoxal with benzene:[16]
As usual, the aldehyde group is more reactive electrophile than the phenone.
Scope and variations
Alkylation of benzene and ethylene, one of the largest scale reactions practiced commercially.
This reaction is related to several classic named reactions:
A reaction of phthalic anhydride with resorcinol in the presence of zinc chloride gives the fluorophore fluorescein. Replacing resorcinol by N,N-diethylaminophenol in this reaction gives rhodamine B:
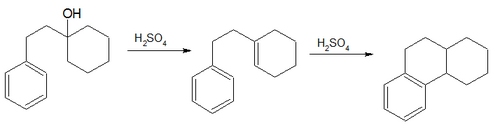
The product formed in this reaction is then analogously reduced, followed by a dehydrogenation reaction (with the reagent SeO2 for example) to extend the aromatic ring system.[28]
Friedel–Crafts test for aromatic hydrocarbons
Reaction of chloroform with aromatic compounds using an aluminium chloride catalyst gives triarylmethanes, which are often brightly colored, as is the case in triarylmethane dyes. This is a bench test for aromatic compounds.[29]
↑ Grzybowski, M.; Skonieczny, K.; Butenschön, H.; Gryko, D. T. (2013). "Comparison of Oxidative Aromatic Coupling and the Scholl Reaction". Angewandte Chemie International Edition. 52 (38): 9900–9930. doi:10.1002/anie.201210238. PMID23852649.
↑ This reaction with phosphorus pentoxide: Kamp, J. V. D.; Mosettig, E. (1936). "Trans- and Cis-As-Octahydrophenanthrene". Journal of the American Chemical Society. 58 (6): 1062–1063. doi:10.1021/ja01297a514.
↑ John C. Gilbert., Stephen F. Martin. Brooks/Cole CENGAGE Learning, 2011. pp 872. 25.10 Aromatic Hydrocarbons and Aryl Halides – Classification test. ISBN978-1-4390-4914-3
Friedel–Crafts reactions published on Organic Syntheses
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.