Androgen receptor antagonistic potency of spironolactone, cyproterone acetate, and flutamide in castrated male rats treated with exogenous testosterone (as measured by inhibition of androgen-dependent ventral prostate weight). Bicalutamide is a much more potent androgen receptor antagonist than flutamide both in animals and in humans.
Bicalutamide acts as a highly selectivecompetitivesilent antagonist of the androgen receptor (AR) (IC50Tooltip half-maximal inhibitory concentration = 159–243nM), the major biological target of the androgen sex hormones testosterone and dihydrotestosterone (DHT).[21][22][23][24] It has no capacity to activate the AR under normal physiological circumstances.[25] In addition to competitive antagonism of the AR, bicalutamide has been found to accelerate the degradation of the AR, and this action may also be involved in its activity as an antiandrogen.[26] The activity of bicalutamide lies in the (R)-isomer, which binds to the AR with an affinity that is about 30-fold higher than that of the (S)-isomer.[27] Levels of the (R)-isomer also notably are 100-fold higher than those of the (S)-isomer at steady-state.[28][29] In terms of relative binding affinity (RBA) for the AR, bicalutamide has shown 0.29 to 6.4%, hydroxyflutamide 0.20 to 1%, flutamide <0.0057%, nilutamide 0.9%, and cyproterone acetate 2.2 to 7.8% of that of metribolone (100%) or DHT (100%) in different studies.[15][30][31]
In relation to its selectivity for the AR, unlike steroidal antiandrogens (SAAs) such as CPA and megestrol acetate (MGA), bicalutamide does not interact importantly with other steroid hormone receptors (including the ERsTooltip estrogen receptors, PRsTooltip progesterone receptors, GRTooltip glucocorticoid receptor, or MRTooltip mineralocorticoid receptor), and in accordance, has no clinically relevant additional, off-target hormonal activity (estrogenic or antiestrogenic, progestogenic or antiprogestogenic, glucocorticoid or antiglucocorticoid, or mineralocorticoid or antimineralocorticoid).[32][18][27][33] However, it has been reported that bicalutamide has weak affinity for the progesterone receptor (PR) (~100- to 500-fold lower than for the AR),[11][12][13][9] where it acts as an antagonist (with only ~12-fold lower functional inhibition relative to the AR in one study).[9][34] Hence, bicalutamide may have some antiprogestogenic activity, although the clinical relevance of this is unknown.[9][34] Bicalutamide does not inhibit 5α-reductase and is not known to inhibit other enzymes involved in androgen steroidogenesis (e.g., CYP17A1).[25] Although bicalutamide does not bind to the ERs, it can increase estrogen levels secondarily to blockade of the AR when used as a monotherapy in males, and for this reason, the medication can indirectly activate the ERs to a degree and hence have some indirect estrogenic effects in men.[35] Also in contrast to SAAs, bicalutamide neither inhibits nor suppresses androgen production in the body (i.e., it does not act as an antigonadotropin or steroidogenesis inhibitor),[2] and instead exclusively mediates its antiandrogen effects by blocking androgen binding and subsequent receptor activation at the level of the AR.[32][27]
In addition to the classical nuclear AR, bicalutamide has also been identified as a potent antagonist of ZIP9, a membrane androgen receptor (mAR) and zinc transporter protein, with an IC50 of 66.3nM (relative to Kd = 17.9nM for testosterone).[36] This protein appears to be involved in prostate cancer and breast cancer.[37] Bicalutamide failed to affect testosterone signaling mediated by GPRC6A, another mAR, on the other hand.[38]
Footnotes:a = Controversial data. Sources: See template.
Drug levels, androgen levels, and efficacy
The affinity of bicalutamide for the AR is approximately 30 to 100times lower than that of DHT (IC50 ≈ 3.8nM), the main endogenousligand of the receptor in the prostate gland.[45][24][1][46] However, sufficiently high relative concentrations of bicalutamide (1,000- to 10,000-fold excess) are able to completely prevent activation of the AR by androgens like DHT and testosterone and subsequent upregulation of the transcription of androgen-responsive genes and associated effects.[47][18][17][48] At steady-state, relative to the normal adult male range for testosterone levels (300–1,000ng/dL),[49] circulating concentrations of bicalutamide at 50mg/day are roughly 600 to 2,500times higher and at 150mg/day around 1,500 to 8,000times higher than circulating testosterone levels, while bicalutamide concentrations, relative to the mean testosterone levels present in men who have been surgically castrated (15ng/dL),[50] are approximately 42,000times higher than testosterone levels at 50mg/day.[51][52][53][54][55]
Median PSA reduction (%) with bicalutamide monotherapy at different dosages (mg/day) and with castration monotherapy.
Whereas testosterone is the major circulating androgen, DHT is the major androgen in the prostate gland.[57]DHT levels in circulation are relatively low and only approximately 10% of those of circulating testosterone levels.[57] Conversely, local concentrations of DHT in the prostate gland are 8- to 10-fold higher than circulating levels of DHT.[58][32] This is due to high expression of 5α-reductase in the prostate gland, which very efficiently catalyzes the formation of DHT from testosterone[58] such that over 90% of intraprostatic testosterone is converted into DHT.[59][60] Relative to testosterone, DHT is 2.5- to 10-fold as potent as an AR agonist in bioassays, and hence, is a much stronger androgen in comparison.[61] As such, AR signaling is exceptionally high in the prostate gland, and the effectiveness of bicalutamide monotherapy in the treatment of prostate cancer, which is roughly equivalent to that of gonadotropin-releasing hormone analogues (GnRH analogues),[62][48][63][64] demonstrates a capacity of bicalutamide to strongly antagonize the AR. On the other hand, GnRH analogue monotherapy achieves only a 50 to 60% reduction in levels of DHT in the prostate gland,[24][65] and combined androgen blockade (CAB), the combination of surgical castration or a GnRH analogue and bicalutamide, is significantly more effective than either modality alone in the treatment of prostate cancer.[21][66] Bicalutamide monotherapy has been found to decrease circulating levels of prostate-specific antigen (PSA), a marker of prostate cancer growth, by 57% at 10mg/day, 73% at 30mg/day, 90% at 50mg/day, 97% at 100mg/day, and 97% at 150mg/day, while a 97% reduction in PSA is observed with 50mg/day bicalutamide as a part of CAB.[24] It has also been reported that bicalutamide monotherapy decreases median circulating levels of PSA at 3months by 86.7% at 100mg/day, 91.1% at 150mg/day, and 93.8% at 200mg/day (relative to 94–97% for castration).[56] Above a bicalutamide monotherapy dosage of 200mg/day, up to 600mg/day, decreases in PSA levels reach a plateau.[34][56] In a study of very-high-dose bicalutamide monotherapy, decreases in PSA levels after 12weeks were approximately 93% with 300mg/day, 96% with 450mg/day, 96% with 600mg/day, and 96% with castration.[67]
Earlier studies with bicalutamide instead assessed changes in prostatic acid phosphatase (PAP) levels; proportions of patients with decreases of PAP of greater than or equal to 50% were 33% with 10mg/day, 53% with 30mg/day, and 83% with 50mg/day bicalutamide.[68]PSA is a more sensitive and specific prostate cancer tumor marker than PAP and subsequent studies employed PSA.[68]
Despite the high medication levels that are achieved, due to their relatively low affinities for the AR, it has been suggested that 5 to 10% of DHT may remain unblocked in the prostate gland with CAB using standard doses of first-generation NSAAs.[69][70][71] In accordance, second-generation NSAAs like enzalutamide and apalutamide, which have 5- to 10-fold higher affinity for the AR than bicalutamide, have been found to be more effective than bicalutamide in the treatment of prostate cancer.[70] However, in the TERRAIN and STRIVE trials, which compared bicalutamide and enzalutamide as a component of CAB and found that enzalutamide extended life by 3 to 4times as much time as bicalutamide, the dosage of enzalutamide used (160mg) was over 3times that of the dosage of bicalutamide used (50mg).[72][73] As a result, it has been suggested that the 50mg/day dosage of bicalutamide used in this study and in CAB in general may be suboptimal.[74][34][73] This is in accordance with clinical findings that PSA decreases with CAB using bicalutamide plateau at a dosage of bicalutamide of 150 to 200mg/day.[34]
In women, total testosterone levels are 20-fold and free testosterone levels 40-fold lower relative to men.[75] In addition, whereas bicalutamide monotherapy can increase testosterone levels by up to 2-fold in men,[64][76] the medication does not increase testosterone levels in women.[77][78][79] For these reasons, much lower dosages of bicalutamide (e.g., 25mg/day in the hirsutism studies) may be used in women with significant antiandrogenic effectiveness.[80][81][82][83]
Footnotes:a = After 29 to 85 days of treatment. Sources:[85]
In men, blockade of the AR by bicalutamide in the pituitary gland and hypothalamus prevents the negative feedback of androgens on the hypothalamic–pituitary–gonadal (HPG) axis, resulting in an increase in luteinizing hormone (LH) secretion and levels.[86]Follicle-stimulating hormone (FSH) levels, in contrast, remain essentially unchanged.[87] The increase in LH levels leads to an elevation in androgen and estrogen levels.[88] At a dosage of 150mg/day, bicalutamide has been found to increase testosterone levels by about 1.5- to 2-fold (59–97% increase) and estradiol levels by about 1.5- to 2.5-fold (65–146% increase).[64][76][83] However, there is variability in these increases, and in one clinical trial, the highest testosterone concentration that occurred with 50mg/day bicalutamide monotherapy was 2,306ng/dL, which was five times the normal average level of testosterone in men.[42][89] Levels of DHT are also increased to a lesser extent (by 24–30%), and concentrations of sex hormone-binding globulin (SHBG) and prolactin increase as well (by 8–42% and 40–65%, respectively) secondary to the increase in estradiol levels.[83][90][91] The estradiol concentrations produced in men by bicalutamide monotherapy are said to approximate the low-normal estradiol levels of a premenopausal woman,[76] while testosterone levels generally remain in the high end of the normal male range and rarely exceed it.[92][32] Dosages of bicalutamide of 10mg, 30mg, and 50mg per day have been found to produce a "moderate" effect on sex hormone levels in men with prostate cancer (notably providing indication that the drug has clinically-relevant antiandrogen effects in males at a dosage as low as 10mg/day).[93][85] The elevated levels of gonadotropins and gonadal steroids associated with NSAA monotherapy is a unique endocrine state which can be described as "hypergonadotropic hypergonadism".[94][95]
Bicalutamide increases androgen and estrogen levels only in men, and does not do so in women.[77][78][79] This is because androgen levels are comparatively far lower in women and in turn exert little to no basal suppression of the HPG axis.[79] Minimal or no changes of importance in levels of total testosterone, free testosterone, dihydrotestosterone, estradiol, androstenedione (A4), dehydroepiandrosterone (DHEA), dehydroepiandrosterone sulfate (DHEA-S), 3α-androstanediol glucuronide (3α-ADG), progesterone, 17α-hydroxyprogesterone (17α-OHP), LH, FSH, prolactin, or SHBG have been observed in women with hirsutism with or without polycystic ovary syndrome that were treated with 25 or 50mg/day bicalutamide for 6 to 12months.[96][97] However, in one study in women with polycystic ovary syndrome, 25mg/day bicalutamide significantly decreased levels of total and free testosterone and significantly increased levels of SHBG.[98] In addition to the minimal changes in hormone levels in women, although bicalutamide monotherapy increases gonadotropin and sex hormone levels in men, this will not occur if bicalutamide is combined with an antigonadotropin such as a GnRH analogue, estrogen, or progestogen, as these medications maintain negative feedback on the HPG axis.[25][99][100][101]
The reason that testosterone levels are elevated but almost always remain in the normal male range with bicalutamide monotherapy is thought to be due to the concomitantly increased levels of estradiol, as estradiol is potently antigonadotropic and limits secretion of LH.[86] In fact, estradiol is a much stronger inhibitor of gonadotropin secretion than is testosterone, and even though circulating concentrations of estradiol are far lower than those of testosterone in men, it is said that estradiol is nonetheless likely the major feedback regulator of gonadotropin secretion in this sex.[102] In accordance, clomifene, a selective estrogen receptor modulator with antiestrogenic activity, has been found to increase testosterone levels to as much as 250% of initial values in men with hypogonadism,[103] and a study of clomifene treatment in normal men observed increases in FSH and LH levels of 70–360% and 200–700%, respectively, with increases in testosterone levels that were similar to the increases seen with the gonadotropins.[104][105] In addition to systemic or circulating estradiol, local aromatization of testosterone into estradiol in the hypothalamus and pituitary gland may contribute to suppression of gonadotropin secretion.[102]
Bicalutamide more than blocks the effects of the increased testosterone levels that it induces in men, which is evidenced by its dose-dependent antiandrogenic effects (e.g., PSA decreases) and by the fact that monotherapy with the drug is about as effective as GnRH analogue therapy in the treatment of prostate cancer.[85][62] However, in contrast, the effects of the elevated estrogen levels remain unopposed by bicalutamide, and this is importantly involved in the feminizing side effects (e.g., gynecomastia) of the drug in men.[106]
Testosterone levels decline with age in men and younger men have higher testosterone levels on average than older men.[107] Men with prostate cancer treated with bicalutamide are relatively elderly.[108] The increases in testosterone levels with NSAAs like flutamide and bicalutamide may result in greater absolute levels of testosterone and estradiol in younger men than in older men.[109] In one study that administered flutamide, free testosterone levels in young men increased from about 26pg/mL at baseline to about 34pg/mL with flutamide (+31%) and in elderly men from about 16pg/mL at baseline to about 21pg/mL (+31%) with flutamide.[109] Hence, free testosterone levels with flutamide were approximately 1.6-fold higher in young men than in elderly men in this study.[109] In the case of estradiol, total estradiol levels in young men increased from about 26pg/mL at baseline to about 45pg/mL with flutamide (+73%) and in elderly men changed from about 31pg/mL to about 30pg/mL (–3%).[109] Other studies have similarly found relatively high absolute testosterone levels with NSAAs in young males.[110][111][112] For instance, one study administering flutamide to late-pubertal males found that total testosterone levels increased from 729ng/dL at baseline to 991ng/dL with flutamide (+34%).[111][112]
Changes in hormone levels with bicalutamide monotherapy
Testosterone levels with 10, 30, and 50mg/day bicalutamide monotherapy in men with prostate cancer.[89]
Testosterone levels with 10 to 200mg/day bicalutamide monotherapy in men with prostate cancer.[85]
Estradiol levels with 10, 30, and 50mg/day bicalutamide monotherapy in men with prostate cancer.[89]
Estradiol levels with 10 to 200mg/day bicalutamide monotherapy in men with prostate cancer.[85]
Differences from castration
It has been proposed that the increase in estrogen levels caused by NSAAs like bicalutamide compensates for androgen blockade in the brain, which may explain differences in the side effect profiles of these drugs relative to GnRH analogues/castration, combined androgen blockade, and CPA (which, in contrast, decrease both androgen and estrogen levels).[113][114][115] In the case of sexual interest and function, this notion is supported by a variety of findings including animal studies showing that estrogen deficiency results in diminished sexual behavior, treatment with tamoxifen resulting in significantly lowered libido in 30% of men receiving it for male breast cancer, and estrogen administration restoring libido and the frequency of sexual intercourse in men with congenital estrogen deficiency, among others.[113][114][115][116]
Several metabolites of testosterone and DHT, including estradiol, 3α-androstanediol, and 3β-androstanediol, are estrogens (mainly potent ERβTooltip estrogen receptor beta agonists in the cases of the latter two), and 3α-androstanediol is additionally a potent GABAA receptor-potentiating neurosteroid.[117][118] Due to the fact that bicalutamide does not lower testosterone levels, the levels of these metabolites would not be expected to be lowered either, unlike with therapies such as GnRH analogues. (Indeed, testosterone, DHT, and estradiol levels are actually raised by bicalutamide treatment, and for this reason, levels of 3α- and 3β-androstanediol might be elevated to some degree similarly.) These metabolites of testosterone have been found to have AR-independent positive effects on sexual motivation,[117][118][119][120] and may be involved in the preservation of sexual interest and function by bicalutamide and other NSAAs.[113] However, a study found that a combination of bicalutamide and dutasteride, a 5α-reductase inhibitor and inhibitor of neurosteroid biosynthesis, produced fewer sexual side effects than GnRH analogue therapy, specifically suggesting the role of estradiol in the preservation sexual interest and function with bicalutamide monotherapy rather than of DHT metabolites.[121]
As an alternative possibility to testosterone metabolites preserving sexual desire and function with bicalutamide, it has been suggested that bicalutamide may not be able to block the actions of androgens in the brain to a degree sufficient to cause substantial sexual impairment.[122]
Although bicalutamide has been characterized as a silent antagonist of the AR in prostate tissue and other contexts, and despite its overall antiandrogenic profile, there is evidence that the drug may activate the AR to some degree in certain other tissues, such as muscle and bone among others.[123][124][125] This would make bicalutamide a selective androgen receptor modulator (SARM), or a tissue-selective mixed agonist/antagonist or partial agonist of the AR, with antiandrogenic effects in some tissues and androgenic effects in other tissues, rather than a pure antiandrogen.[123][124][125][23][126] This would be similar to certain other SARMs structurally related to bicalutamide, like enobosarm, which in animals have potent anabolic effects in muscle and bone but show partially agonistic or antagonistic effects in the prostate or seminal vesicles.[127][128]
Bicalutamide has been found to not oppose testosterone-induced increases in levator animuscle weight in immature castrated male rats at doses of the drug (e.g., 0.08–2mg/kg) that reduce or even completely block testosterone-induced prostate gland and seminal vesicle growth.[18][17][129] In other studies, higher doses of bicalutamide (e.g., 10–30mg/kg) reduced levator ani muscle weight (by ~40% or more) in gonadally intact male rats but had no effect on lean body mass (a surrogate of muscle mass), whereas castration reduced levator ani weight by around 70% and lean body mass by around 25%.[130][131] Moreover, not only has bicalutamide been found not to reduce levator ani weight or lean body mass in male rats at doses that are potently antiandrogenic in the prostate, the drug has been found to partially antagonize castration-induced body weightloss and lean body mass loss in male rats.[132] This effect also occurred with JNJ-26146900, a SARM, and with DHT, although neither JNJ-26146900 nor bicalutamide were as effective as DHT at preserving body weight or lean mass.[132] As such, on the basis of such preclinical findings, it has been said that bicalutamide does not have marked antianabolic effects in muscle, and hence shows tissue selectivity in its antiandrogenic actions.[18][17] Moreover, bicalutamide may also actually activate the AR in muscle to preserve muscle mass.[132] If these findings were to translate to humans, then bicalutamide would inhibit prostate cancer growth without the antiandrogenic muscle atrophy that occurs with castration.[18][17] Analogously to the animal findings, high-dose bicalutamide monotherapy has been found to preserve lean muscle mass and muscle strength in men with prostate cancer relative to GnRH agonists.[91][133][134]
Androgens are known to exert direct effects on bone remodeling through activation of ARs in osteoblasts and osteoclasts.[124] In relation to this, androgen deprivation therapy for prostate cancer has been associated with osteopenia.[124] It has been found that bicalutamide monotherapy, in spite of its antiandrogenic activity, does not affect bone remodeling or bone mineral density in healthy male rats.[124] It was proposed, on the basis of these findings, that bicalutamide may act as a SARM for bone remodeling in osteoblasts, different from its antiandrogenic actions in the prostate gland.[124] Likewise, hydroxyflutamide, the active metabolite of the related NSAA flutamide, has been found to inhibit interleukin-6 production in an androgen-responsive osteoblast cell line analogously to androgens, and hence has shown SARM-like activity in bone.[124][135] In accordance with preclinical findings, bicalutamide monotherapy preserves bone mineral density in men with prostate cancer.[54][133] Additionally, clinical studies have reported that combined androgen blockade (CAB) with bicalutamide helps to preserve bone parameters to a greater extent than castration monotherapy.[136][125][137]
It is notable however that in contrast to castration, bicalutamide monotherapy preserves and increases estrogen levels, and estrogens have positive effects both on bone and muscle.[138] This may explain preservation of bone with bicalutamide monotherapy observed in animals and humans.[54][133][124] However, it could not explain findings of improved bone parameters with bicalutamide plus castration relative to castration monotherapy, as estrogens are deprived in this context.[136][125][137] In terms of muscle, castration and menopause reduce muscle mass in women and men, and in women, estradiol replacement therapy prevents loss of lean body mass.[139] Conversely however, estrogen deficiency did not decrease lean body mass in men treated with a GnRH agonist and testosterone with versus without an aromatase inhibitor.[140] In any case, findings that bicalutamide helps to preserve body weight and lean body mass in castrated male rats also could not be explained by preservation of estrogen levels.[132]
Aside from muscle, bicalutamide has also been found to act not as an AR silent antagonist but as a weak partial agonist of the receptor with SARM-like activity in AR-positive MDA-MB-453breast cancercells.[130] Whereas a pure AR antagonist should have no effect, bicalutamide regulated almost 50% of 189 DHT-responsive genes in this cell line and induced these genes around 25% as effectively as the highest tested concentration of DHT.[130] DHT and SARMs like enobosarm and GTx-027 inhibit the proliferation of AR-expressing MDA-MB-231 breast cancer cells, whereas findings for bicalutamide are conflicting.[141][142] Bicalutamide has also shown SARM-like or weak partial agonist activity in certain other in vitro AR bioassays.[130][143][144]
A very high (50mg/kg) dose of bicalutamide has been found to suppress testosterone levels by about 91% in gonadally intact male mice, which was to a similar degree as the SARM MK-4541.[130] These findings suggest that bicalutamide at very high doses may have antigonadotropic effects that may be mediated by SARM-like activity.[130] Conversely however, lower doses of bicalutamide (1–25mg/kg) have not been found to affect testosterone levels in gonadally intact male rats, and similar or higher doses of bicalutamide (20–100mg/kg) did not affect testosterone levels in gonadally intact male dogs.[18] At clinically studied doses in humans (up to 600mg/day), which are much lower on a mg/kg basis than those used in the preceding animal studies, bicalutamide has only shown increases in testosterone levels and no testosterone suppression.[85][67]
There is some indication that the related NSAA nilutamide may also have SARM-like properties in certain tissues, as suggested by stimulation of erythropoiesis in men with prostate cancer.[145] Additionally, hydroxyflutamide, the active metabolite of the related NSAA flutamide, has shown SARM-like activity in an androgen-responsive osteoblast cell line.[135] Similarly, along with CPA, flutamide and hydroxyflutamide, though not bicalutamide, have been reported to act as SARMs or AR partial agonists in prostate cancer cells.[31][146] Novel SARMs like enobosarm, with antiandrogenic effects in the prostate gland like bicalutamide but potent anabolic effects in muscle and bone, have also been developed.[130][126]
Bicalutamide has also been described as a functional "SARM" due to peripheral selectivity and inability to cross into the central nervous system and block ARs in this part of the body, resulting in antiandrogenic action in the periphery and lack of effects in the brain.[147] However, while peripheral selectivity was initially observed in animal studies, bicalutamide did not end up showing peripheral selectivity in humans.[147]
Paradoxical stimulation of late-stage prostate cancer
Though a pure, or silent antagonist of the AR under normal circumstances, bicalutamide, as well as other earlier antiandrogens like flutamide and nilutamide, have been found to possess weak partial agonist properties in the setting of AR overexpression and agonist activity in the case of certain mutations in the ligand-binding domain (LBD) of the AR.[148][149] As both of these circumstances can eventually occur in prostate cancer, resistance to bicalutamide usually develops and the drug has the potential to paradoxically stimulate tumor growth when this happens.[148][150] This is the mechanism of the phenomenon of antiandrogen withdrawal syndrome, where antiandrogen discontinuation paradoxically slows the rate of tumor growth.[150] The newer drug enzalutamide has been shown not to have agonistic properties in the context of overexpression of the AR, though certain mutations in the AR can still convert it from an antagonist to agonist.[148]
A second mechanism of bicalutamide resistance has been shown to be mediated by an interaction between macrophages and cancer cells.[151][152] In a typical scenario, the bicalutamide-AR complex translocates inside the nucleus and binds to androgen response elements (AREs). It then recruits a N-CoR corepressor complex which leads to the repression of androgen receptor target genes by preventing their transcription.[152] The triggering of proinflammatory pathways through IL-1 signaling by macrophages infiltration causes the recruitment of TAB2 as a component of the N-CoR complex. TAB2 in turn serves as a beacon for the recruitment of MEKK1 which then dismisses the N-CoR complex and derepresses its target gene, in effect converting bicalutamide into an AR agonist.[151] This mechanism does not rely on mutations in the LBD of the AR.
Induction of breast development
Bicalutamide monotherapy and breast side effects in dose-ranging studies in men
Footnotes:a = Testosterone levels increased to ~460–610ng/dL and estradiol levels to ~32–51pg/mL. b = Testosterone levels increased to ~505–715ng/dL and estradiol levels to ~32–53pg/mL. c = Testosterone levels increased to ~540–600ng/dL and estradiol levels to ~29–34pg/mL.
In transgender women, breast development is a desired effect of antiandrogen and/or estrogen treatment.[156][157] Bicalutamide induces breast development in individuals assigned male at birth by two mechanisms: 1) blocking androgen signaling in breast tissue; and 2) increasing estrogen levels.[158][159][160] Estrogen is responsible for the induction of breast development under normal circumstances, while androgens powerfully suppress estrogen-induced breast growth.[161][162] It has been found that very low levels of estrogen can induce breast development in the presence of low or no androgen signaling.[161][163][164] In accordance, not only does bicalutamide induce gynecomastia at a high rate when given as a monotherapy to men with prostate cancer (38–85%; 66% in one very large trial),[160][158] similarly to high-dose estrogen therapy with diethylstilbestrol (41–77%), NSAAs have been found to result in a higher incidence of gynecomastia in combination with a GnRH analogue (13–25%) relative to GnRH analogue therapy or castration alone (1–16%) (in spite of the presence of only castrate levels of estrogen in both cases).[165][35][166][56] The rate of gynecomastia with CAB is also higher than with CPA monotherapy (7%).[165]
A study of men treated with NSAA (flutamide or bicalutamide) monotherapy for prostate cancer found that NSAAs induced full ductal development and moderate lobuloalveolar development of the breasts from a histological standpoint.[167][168][169] The study also found that, in contrast, treatment of transgender women with estrogen and CPA (which is progestogenic in addition to antiandrogenic, unlike NSAAs) resulted in full lobuloalevolar development, as well as pregnancy-like breast hyperplasia in two of the subjects.[167][169] In addition, it was observed that the lobuloalveolar maturation reversed upon discontinuation of CPA after sex reassignment surgery in these individuals.[167] It was concluded that progestogen in addition to antiandrogen/estrogen treatment is required for the induction of full female histological breast development (i.e., that includes complete lobuloalveolar maturation), and that continued progestogen treatment is necessary to maintain such maturation.[167][168] It should be noted however that although these findings may have important implications in the contexts of lactation and breastfeeding, epithelial tissue accounts for approximately only 10% of breast volume (with the bulk of the breasts (80–90%) being represented by stromal or adipose tissue),[170][171][172][173] and it is uncertain to what extent, if any, that development of lobuloalveolar structures (a form of epithelial tissue) contributes to breast size and/or shape.[156]
Effects on spermatogenesis and fertility
Spermatogenesis and male fertility are dependent on FSH, LH, and high levels of testosterone within the testicles.[58][174]LH does not seem to be involved in spermatogenesis outside of its role in inducing production of testosterone by the Leydig cells in the seminiferous tubules (which make up approximately 80%[175] of the bulk of the testes),[176] whereas this is not the case for FSH, which is importantly involved.[177][178] In accordance with the fact that the testes are the source of 95% of circulating testosterone in the body, local levels of testosterone inside of the testes are extremely high, ranging from 20- to 200-fold higher than circulating concentrations.[179][60] Moreover, high levels of testosterone within the testes are required for spermatogenesis,[174] although only a small fraction (5–10%) of normal levels appears to actually be necessary for spermatogenesis.[60][180]
Unlike with antigonadotropic antiandrogens like CPA and GnRH analogues, it has been reported that bicalutamide monotherapy (at 50mg/day) has very little or no effect on the ultrastructure of the testes and on spermatogenesis in men even after long-term therapy (>4years).[181][182] This may be explained by the extremely high local levels of testosterone in the testes, in that it is likely that systemic bicalutamide therapy is unable to achieve concentrations of the drug within the testes that are able to considerably block androgen signaling in this part of the body.[181] This is particularly so considering that bicalutamide increases circulating testosterone levels, and by extension gonadal testosterone production, by up to two-fold in males,[88] and that only a small fraction of normal intratesticular testosterone levels, and by extension androgen action, appears to be necessary to maintain spermatogenesis.[60][180] Bicalutamide monotherapy at 50mg/day causes no or clinically unimportant Leydig cell hyperplasia.[182][183][184]
In contrast to bicalutamide and other pure antiandrogens or NSAAs, antigonadotropic antiandrogens suppress gonadotropin secretion, which in turn diminishes testosterone production by the testes as well as the maintenance of the testes by FSH, resulting in atrophy and loss of their function.[185] As such, bicalutamide and other NSAAs may uniquely have the potential to preserve testicular function and spermatogenesis and thus male fertility relative to alternative therapies.[181][186] In accordance with this notion, a study found that prolonged, high-dose bicalutamide treatment had minimal effects on fertility in male rats.[187] However, another study found that low-dose bicalutamide administration resulted in testicular atrophy and reduced the germ cell count in the testes of male rats by almost 50%, though the rate of successful fertilization and pregnancy following mating was not assessed.[188] Additional studies found that bicalutamide decreased testes weights, altered testes histology, and decreased sperm count in male rats.[189][190][191] Yet another study found that bicalutamide has no effect on testes weights or spermatogenesis in male rats.[192]
Treatment of men with exogenous testosterone or other AAS results in suppression of gonadotropin secretion and gonadal testosterone production due to their antigonadotropic effects or activation of the AR in the pituitary gland, resulting in inhibition or abolition of spermatogenesis and fertility:[193]
Treatment of an infertile man with testosterone does [not] improve spermatogenesis, since exogenous administrated testosterone and its metabolite estrogen will suppress both GnRH production by the hypothalamus and luteinizing hormone production by the pituitary gland and subsequently suppress testicular testosterone production. Also, high levels of testosterone are needed inside the testis and this can never be accomplished by oral or parenteral administration of androgens. Suppression of testosterone production by the leydig cells will result in a deficient spermatogenesis, as can be seen in men taking anabolic–androgenic steroids.[193]
In contrast, pure AR antagonists would, in theory, result in the opposite (although reduced semen volume and sexual dysfunction may occur):[194]
It is theoretically a sound hypothesis that the spermatogenesis can be increased by indirectly stimulating FSH and LH secretions from the pituitary gland. However, for this to fructify, it requires the use of testosterone antagonist to nullify the negative feedback effect of circulating testosterone on the release of FSH and LH, thus augmenting the secretion of testosterone and spermatogenesis. Unfortunately, a testosterone antagonist will be unacceptable to males, as it may reduce secondary sexual functions including erection and ejaculation that is vital for the successful fertilization.[194]
However, while bicalutamide does not appear to adversely influence testicular spermatogenesis, and healthy sperm can be produced within the testes during bicalutamide monotherapy, AR antagonists may be able to interfere with male fertility via interference with androgen signaling beyond the testes.[195] The maturation as well as transport of sperm occurs not only in the testes but also outside of the testes in the epididymides and vas deferens, and these processes in these tissues are dependent on AR signaling similarly to testicular spermatogenesis.[195] However, whereas androgen levels are extremely high in the testes, this is not true in the epididymides and vas deferens.[195] As androgen levels are relatively low in these tissues, at least compared to the testes, bicalutamide may be able to block AR signaling in these parts of the body to an extent that is sufficient to interfere with male fertility.[195] Indeed, the AAS mesterolone has been used to improve sperm quality and fertility in men because, apparently unlike other AAS, it shows minimal antigonadotropic effects at typical clinical dosages but activates the AR and thereby supports sperm maturation in the epididymides.[196] However, this use of mesterolone is controversial and its efficacy for such purposes is not fully certain.[196]
Although bicalutamide alone would appear to have minimal detrimental effect on testicular spermatogenesis and hence on certain aspects of male fertility, other hormonal agents that bicalutamide may be combined with, including GnRH analogues and particularly estrogens (as in transgender hormone therapy), can have a considerable detrimental effect on fertility.[197][198] This is largely a consequence of their antigonadotropic activity.[198] Antigonadotropic agents like high-dose CPA,[199][200] high-dose androgens (e.g., testosterone esters), and GnRH antagonists (though notably not GnRH agonists in the case of fertility) produce hypogonadism and high rates of severe or complete infertility (e.g., severe oligospermia or complete azoospermia) in men.[198] However, these effects are fully and often rapidly reversible with their discontinuation, even after prolonged treatment.[198][200] In contrast, while estrogens at sufficiently high dosages similarly are able to produce hypogonadism and to abolish or severely impair spermatogenesis,[197] this is not necessarily reversible in the case of estrogens and can be long-lasting after prolonged exposure.[198][201] The difference is attributed to an apparently unique, direct cytotoxic and adverse effect of high concentrations of estrogens on the Leydig cells of the testes.[198][201]
Other activities
Cytochrome P450 modulation
It has been reported that bicalutamide may have the potential to inhibit the enzymesCYP3A4 and, to a lesser extent, CYP2C9, CYP2C19, and CYP2D6, based on in vitro research.[1] However, no relevant inhibition of CYP3A4 has been observed in vivo with bicalutamide at a dose of 150mg (using midazolam as a specific marker of CYP3A4 activity).[1] In animals, bicalutamide has been found to be an inducer of certain cytochrome P450 enzymes.[1] However, dosages of 150mg/day or less have shown no evidence of this in humans.[1]
Bicalutamide has been identified as a strong CYP27A1 (cholesterol 27-hydroxylase) inhibitor in vitro.[202] CYP27A1 converts cholesterol into 27-hydroxycholesterol, an oxysterol that has multiple biological functions including direct, tissue-specific activation of the ER (it has been characterized as a selective estrogen receptor modulator) and the liver X receptor.[202] 27-Hydroxycholesterol has been found to increase ER-positive breast cancer cell growth via its estrogenic action, and hence, it has been proposed that bicalutamide and other CYP27A1 inhibitors may be effective as adjuvant therapies to aromatase inhibitors in the treatment of ER-positive breast cancer.[202] In addition to CYP27A1, bicalutamide has been found to bind to and inhibit CYP46A1 (cholesterol 24-hydroxylase) in vitro, but this has yet to be assessed and confirmed in vivo.[203]
All of the NSAAs approved for the treatment of prostate cancer have been found to possess an off-target action of acting as weak non-competitive inhibitors of human GABAA receptor currents in vitro to varying extents.[207][208] The IC50 values are 44 μM for flutamide (as hydroxyflutamide), 21 μM for nilutamide, 5.2 μM for bicalutamide, and 3.6 μM for enzalutamide.[207] In addition, flutamide, nilutamide, and enzalutamide have been found to cause convulsions and/or death in mice at sufficiently high doses.[207] Bicalutamide was notably not found to do this, but this was likely simply due to the limited central nervous system penetration of bicalutamide in this species.[207] In any case, enzalutamide is the only approved NSAA that has been found to be associated with a significantly increased incidence of seizures and other associated side effects clinically, so the relevance of the aforementioned findings with regard to bicalutamide and the other NSAAs is unclear.[207]
Bicalutamide is extensively and well-absorbed following oral administration,[1] and its absorption is not affected by food.[2][211] The absolute bioavailability of bicalutamide in humans is unknown due to its very low water solubility and hence lack of an assessable intravenous formulation.[1][2] However, the absolute bioavailability of bicalutamide has been found to be high in animals at low doses (109% in mice at 10mg/kg; 72% in rats at 1mg/kg; 100% in dogs at 0.1mg/kg), but diminishes with increasing doses such that the bioavailability of bicalutamide is low at high doses (10% in rats at 250mg/kg; 31% in dogs at 100mg/kg).[1][212][213] In accordance, absorption of (R)-bicalutamide in humans is slow and extensive but saturable,[54] with steady-state levels increasing linearly at a dosage of up to 150mg/day and non-linearly at higher dosages.[1]
At higher dosages of 100 to 200mg/day, absorption of bicalutamide is approximately linear, with a small but increasing departure from linearity above 150mg/day.[214] In terms of geometric mean steady-state concentrations of (R)-bicalutamide, the departures from linearity were 4%, 13%, 17%, and 32% with dosages of 100, 150, 200, and 300mg/day, respectively.[1] There is a plateau in steady-state levels of (R)-bicalutamide with bicalutamide dosages above 300mg/day, and, accordingly, dosages of bicalutamide of 300 to 600mg/day result in similar circulating concentrations of (R)-bicalutamide and similar degrees clinically of efficacy, tolerability, and toxicity.[1][67] Relative to 150mg/day bicalutamide, levels of (R)-bicalutamide are about 15% higher at a dosage of 200mg/day and about 50% higher at a dosage of 300mg/day.[67] In contrast to (R)-bicalutamide, the inactive enantiomer (S)-bicalutamide is much more rapidly absorbed (as well as cleared from circulation).[1]
Steady-state concentrations of the drug are reached after 4 to 12 weeks of administration independently of dosage, with an approximate 10- to 20-fold progressive accumulation of circulating levels of (R)-bicalutamide.[54][108][87][53] The relatively long time to reach steady-state is a product of the long elimination half-life of bicalutamide.[53] With single 50mg and 150mg doses of bicalutamide, mean peak concentrations (Cmax) of (R)-bicalutamide are 0.77μg/mL (1.8μmol/L) (at 31 hours) and 1.4μg/mL (3.3μmol/L) (at 39 hours), respectively.[54][51] At steady-state, mean circulating concentrations (Css) of (R)-bicalutamide with 50mg/day and 150mg/day bicalutamide are 8.85μg/mL (20.6μmol/L) and 21.6μg/mL (50.2μmol/L), respectively.[54][51][52][53] In another 150mg/day bicalutamide study, mean circulating concentrations of (R)-bicalutamide were 19.4μg/mL (45.1μmol/L) and 28.5μg/mL (66.3μmol/L) on days 28 and 84 (weeks 4 and 12) of treatment, respectively.[55]
There is wide interindividual variability, up to 15.7-fold, in steady-state (R)-bicalutamide levels with bicalutamide therapy.[1] This is the case for all dosage levels of bicalutamide, and ranges in (R)-bicalutamide levels for different dosages show significant overlap.[1]
The tissuedistribution of bicalutamide is not well-characterized.[216] However, it has been reported that distribution studies with bicalutamide have shown that preferential (i.e., tissue-selective) accumulation in anabolic (e.g., muscle) tissues does not occur.[217] There are no available data on hepatic bicalutamide concentrations in humans, but a rat study found that oral bicalutamide treatment resulted in 4-fold higher concentrations of the drug in the liver relative to plasma (a common finding with orally administered drugs, due to transfer through the hepatic portal system prior to reaching circulation).[1][218] In men receiving 150mg/day bicalutamide, concentrations of (R)-bicalutamide in semen were 4.9μg/mL (11μmol/L), and the amount of the drug that could potentially be delivered to a female partner during sexual intercourse is regarded as low (estimated at 0.3μg/kg) and below the amount that is required to induce changes in the offspring of laboratory animals.[52]
Based on animal research, it was initially thought that bicalutamide was unable to cross the blood–brain barrier into the central nervous system and hence would be a peripherally-selective antiandrogen in humans.[219][18] This conclusion was drawn from the finding that bicalutamide reportedly does not increase LH or testosterone levels in multiple tested animal species, including rats and dogs.[219][18][220][221]AR antagonists like flutamide normally do this by blocking ARs in the pituitary gland and hypothalamus in the brain and thereby disinhibiting the HPG axis.[86] The notion that bicalutamide does not cross the blood–brain barrier is in accordance with rodent tissue distribution studies, which have found low levels of bicalutamide in the hypothalamus and cerebral cortex relative to most peripheral tissues and the pituitary gland (a part of the brain that is outside of the blood–brain barrier).[222][18][223] In spite of the preceding studies however, other rodent studies have contradicted these results and found that bicalutamide does significantly and dose-dependently increase LH and testosterone levels, and in some studies to a similar extent as flutamide.[224][42][9] In any case, in humans, bicalutamide has consistently been found to increase LH and testosterone levels, and to a comparable extent relative to flutamide and nilutamide.[1][86][225][226][42] This occurs to a significant extent at even a very low dosage of 10mg/day bicalutamide.[85] As such, it appears that there may be species differences in the central penetration of bicalutamide and that the medication does indeed cross the blood–brain barrier and affect central function in humans.[1][226] This is supported by potential side effects of bicalutamide, in spite of increased testosterone levels, like hot flashes and decreased sexual interest in men.[227] However, a clinical study comparing bicalutamide and flutamide in men found that bicalutamide had less influence on the HPG axis than flutamide, suggesting that bicalutamide might have a limited degree of peripheral selectivity,[220] at least compared to other NSAAs, in humans.[228]
The metabolism of bicalutamide is hepatic and stereoselective.[3][211] The inactive (S)-enantiomer is metabolized mainly by glucuronidation and is rapidly cleared from circulation, while the active (R)-isomer is slowly hydroxylated and then glucuronidated.[211] In accordance, the active (R)-enantiomer has a far longer elimination half-life than the (S)-isomer,[27] and circulating levels of (R)-bicalutamide are 10- to 20-fold[54] and 100-fold higher than those of (S)-bicalutamide after a single dose and at steady-state, respectively.[28][29] (R)-Bicalutamide is almost exclusively metabolized via hydroxylation into (R)-hydroxybicalutamide by the cytochrome P450enzymeCYP3A4.[1][3][28] Bicalutamide is also glucuronidated by UGT1A9, a UDP-glucuronyltransferase,[4] into bicalutamide glucuronide, and (R)-hydroxybicalutamide glucuronide is formed from the metabolism of (R)-hydroxybicalutamide by UGT1A9.[1][4][6] Similar to the inactive (S)-enantiomer of bicalutamide, (R)-hydroxybicalutamide is glucuronidated and rapidly cleared from circulation.[234] None of the metabolites of bicalutamide are known to be active.[3][5] Following administration of bicalutamide, only low concentrations of the metabolites are detectable in blood plasma, while unchanged bicalutamide predominates.[1] (R)-Bicalutamide has a long elimination half-life of 5.8days with a single dose,[7][92] and an elimination half-life of 7 to 10days with repeated administration, which allows for convenient once-daily dosing of bicalutamide.[8]
This diagram illustrates the primary metabolic pathways involved in the metabolism of bicalutamide in humans.
Elimination
Bicalutamide is eliminated in feces (43%) and urine (34%),[3][211] whereas its metabolites are eliminated in approximately equal proportions in urine and bile.[84][213] It is excreted to a substantial extent in its unmetabolized form, with both bicalutamide and its metabolites excreted mainly as glucuronideconjugates.[27]
Graphs
Bicalutamide levels during therapy with bicalutamide
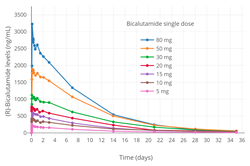
(R)-Bicalutamide levels after a single 5 to 80mg dose of (R)-bicalutamide/HP55S in men.[215] The mean elimination half-life of (R)-bicalutamide in this study was 5.6 to 7.5days.[215]
Bicalutamide levels after a single 50mg dose of bicalutamide in men.[237] The mean elimination half-life of bicalutamide in this study was 4.2days.[237]
Bicalutamide levels after a single 10, 30, or 50mg dose of bicalutamide in men.[238] The mean elimination half-life of bicalutamide in this study was 5.5 to 6.3days.[238]
Mean plasma (R)-bicalutamide concentrations with 10 to 600mg/day bicalutamide in men over the course of 12weeks.[85][67][239]
Steady-state plasma levels of (R)-bicalutamide as a function of bicalutamide dosage (10 to 600mg/day) in men.[85][67][239] Note the divergence from linearity at dosages above 200mg/day, which demonstrates the saturation of absorption with higher dosages of bicalutamide.[239]
1 2 Dole EJ, Holdsworth MT (January 1997). "Nilutamide: an antiandrogen for the treatment of prostate cancer". The Annals of Pharmacotherapy. 31 (1): 65–75. doi:10.1177/106002809703100112. PMID8997470. S2CID20347526. page 67: Currently, information is not available regarding the activity of the major urinary metabolites of bicalutamide, bicalutamide glucuronide, and hydroxybicalutamide glucuronide.
1 2 Schellhammer PF (September 2002). "An evaluation of bicalutamide in the treatment of prostate cancer". Expert Opinion on Pharmacotherapy. 3 (9): 1313–1328. doi:10.1517/14656566.3.9.1313. PMID12186624. S2CID32216411. The clearance of bicalutamide occurs pre- dominantly by hepatic metabolism and glucuronidation, with excretion of the resulting inactive metabolites in the urine and faces.
1 2 3 4 5 Hamann LG, Higuchi RI, Zhi L, Edwards JP, Wang XN, Marschke KB, etal. (February 1998). "Synthesis and biological activity of a novel series of nonsteroidal, peripherally selective androgen receptor antagonists derived from 1,2-dihydropyridono[5,6-g]quinolines". Journal of Medicinal Chemistry. 41 (4): 623–639. doi:10.1021/jm970699s. PMID9484511. Bicalutamide (3) was more potent than 2a in decreasing organ growth in intact animals, due at least in part to its peripheral selectivity, exhibiting only limited penetration of the blood-brain barrier, resulting in at most 2-fold increases in serum LH and T.33
1 2 Nagata N, Miyakawa M, Amano S, Furuya K, Yamamoto N, Nejishima H, etal. (November 2011). "Tetrahydroquinolines as a novel series of nonsteroidal selective androgen receptor modulators: structural requirements for better physicochemical and biological properties". Bioorganic & Medicinal Chemistry Letters. 21 (21): 6310–6313. doi:10.1016/j.bmcl.2011.08.118. PMID21944856.
1 2 Nagata N, Miyakawa M, Amano S, Furuya K, Yamamoto N, Inoguchi K (March 2011). "Design and synthesis of tricyclic tetrahydroquinolines as a new series of nonsteroidal selective androgen receptor modulators (SARMs)". Bioorganic & Medicinal Chemistry Letters. 21 (6): 1744–1747. doi:10.1016/j.bmcl.2011.01.073. PMID21349712.
1 2 Kinoyama I, Taniguchi N, Toyoshima A, Nozawa E, Kamikubo T, Imamura M, etal. (January 2006). "(+)-(2R,5S)-4-[4-cyano-3-(trifluoromethyl)phenyl]-2,5-dimethyl-N-[6-(trifluoromethyl)pyridin-3- yl]piperazine-1-carboxamide (YM580) as an orally potent and peripherally selective nonsteroidal androgen receptor antagonist". Journal of Medicinal Chemistry. 49 (2): 716–726. doi:10.1021/jm050293c. PMID16420057.
1 2 Ayub M, Levell MJ (August 1989). "The effect of ketoconazole related imidazole drugs and antiandrogens on [3H] R 1881 binding to the prostatic androgen receptor and [3H]5 alpha-dihydrotestosterone and [3H]cortisol binding to plasma proteins". J. Steroid Biochem. 33 (2): 251–5. doi:10.1016/0022-4731(89)90301-4. PMID2788775.
↑ Yamasaki K, Sawaki M, Noda S, Muroi T, Takakura S, Mitoma H, etal. (February 2004). "Comparison of the Hershberger assay and androgen receptor binding assay of twelve chemicals". Toxicology. 195 (2–3): 177–186. Bibcode:2004Toxgy.195..177Y. doi:10.1016/j.tox.2003.09.012. PMID14751673.
1 2 3 4 5 Furr BJ (1996). "The development of Casodex (bicalutamide): preclinical studies". European Urology. 29 (2 Suppl 2): 83–95. doi:10.1159/000473846. PMID8717469.
↑ Kolvenbag GJ, Furr BJ, Blackledge GR (December 1998). "Receptor affinity and potency of non-steroidal antiandrogens: translation of preclinical findings into clinical activity". Prostate Cancer and Prostatic Diseases. 1 (6): 307–314. doi:10.1038/sj.pcan.4500262. PMID12496872. S2CID33497597.
1 2 Masiello D, Cheng S, Bubley GJ, Lu ML, Balk SP (July 2002). "Bicalutamide functions as an androgen receptor antagonist by assembly of a transcriptionally inactive receptor". The Journal of Biological Chemistry. 277 (29): 26321–26326. doi:10.1074/jbc.M203310200. PMID12015321. Recent reports demonstrate that the AR can also bind to corepressor proteins, including nuclear corepressor (NCoR), indicating that corepressor binding could further contribute to the in vivo antagonist activity of bicalutamide (42–44). This would suggest down-regulation of a corepressor as another mechanism for development of bicalutamide-resistant PCa. These data also suggest that bicalutamide may function as a selective AR modulator. However, in contrast to the selective estrogen receptor modulators tamoxifen and raloxifene, bicalutamide does not appear to activate the N-terminal activation function (AF-1) of the AR, and cell types or tissues in which bicalutamide functions as an AR agonist have not been identified.
1 2 3 4 Furr BJ (June 1995). "Casodex: preclinical studies and controversies". Annals of the New York Academy of Sciences. 761 (1): 79–96. Bibcode:1995NYASA.761...79F. doi:10.1111/j.1749-6632.1995.tb31371.x. PMID7625752. S2CID37242269. It is suggested that the resulting increase in output of androgen by the testis requires an increased dose of antiandrogen to neutralize any stimulatory effect on the prostate gland. This is a controversial topic [...], but when given in combination with medical or surgical castration, any rise in serum LH becomes irrelevant to clinical outcome.
↑ Waller AS, Sharrard RM, Berthon P, Maitland NJ (June 2000). "Androgen receptor localisation and turnover in human prostate epithelium treated with the antiandrogen, casodex". Journal of Molecular Endocrinology. 24 (3): 339–351. CiteSeerX10.1.1.499.7722. doi:10.1677/jme.0.0240339. PMID10828827.
↑ Payen O, Top S, Vessières A, Brulé E, Lauzier A, Plamont MA, etal. (March 2011). "Synthesis and biological activity of ferrocenyl derivatives of the non-steroidal antiandrogens flutamide and bicalutamide". Journal of Organometallic Chemistry. 696 (5): 1049–1056. doi:10.1016/j.jorganchem.2010.10.051.
1 2 Brooke GN, Gamble SC, Hough MA, Begum S, Dart DA, Odontiadis M, etal. (May 2015). "Antiandrogens act as selective androgen receptor modulators at the proteome level in prostate cancer cells". Mol Cell Proteomics. 14 (5): 1201–16. doi:10.1074/mcp.M113.036764. PMC4424393. PMID25693800. Supplemental Figure 1, Brook et al., Androgens, Dihydrotestosterone (DHT) RBA = 100 (24), Mibolerone (MIB) RBA = 100 (25), Steroidal anti-androgen, Cyproterone Acetate (CPA) RBA = 2.2 (26), Non-steroidal anti-androgen, Bicalutamide (BIC) RBA = 6.36 (24), Hydroxyflutamide (OHF), RBA = 1 (24). Supplemental Figure 1. Chemical structures and relative binding affinities for ligands used. Relative binding affinities are for wild-type AR relative to R1881 [(metribolone)].
1 2 Guise TA, Oefelein MG, Eastham JA, Cookson MS, Higano CS, Smith MR (2007). "Estrogenic side effects of androgen deprivation therapy". Reviews in Urology. 9 (4): 163–180. PMC2213888. PMID18231613. The incidence of gynecomastia varies with the type and duration of ADT.65,83 For example, it is reported in 40% to 80% of men on estrogen therapy (eg, DES), 40% to 70% of men on antiandrogens (bicalutamide, flutamide, or nilutamide, including > 50% with high-dose bicalutamide [150 mg]), 25% of men on combined androgen blockade (LHRH with an antiandrogen), and 10% to 15% of men on LHRH alone or after orchiectomy.66
↑ Thomas P, Converse A, Berg HA (February 2018). "ZIP9, a novel membrane androgen receptor and zinc transporter protein". General and Comparative Endocrinology. 257: 130–136. doi:10.1016/j.ygcen.2017.04.016. PMID28479083.
↑ Furr BJ, Valcaccia B, Curry B, Woodburn JR, Chesterson G, Tucker H (June 1987). "ICI 176,334: a novel non-steroidal, peripherally selective antiandrogen". The Journal of Endocrinology. 113 (3): R7 –R9. doi:10.1677/joe.0.113R007. PMID3625091.
↑ Teutsch G, Goubet F, Battmann T, Bonfils A, Bouchoux F, Cerede E, etal. (January 1994). "Non-steroidal antiandrogens: synthesis and biological profile of high-affinity ligands for the androgen receptor". The Journal of Steroid Biochemistry and Molecular Biology. 48 (1): 111–119. doi:10.1016/0960-0760(94)90257-7. PMID8136296. S2CID31404295.
↑ Winneker RC, Wagner MM, Batzold FH (December 1989). "Studies on the mechanism of action of Win 49596: a steroidal androgen receptor antagonist". Journal of Steroid Biochemistry. 33 (6): 1133–1138. doi:10.1016/0022-4731(89)90420-2. PMID2615358.
1 2 3 4 5 Luo S, Martel C, Leblanc G, Candas B, Singh SM, Labrie C, etal. (1996). "Relative potencies of Flutamide and Casodex: preclinical studies". Endocrine Related Cancer. 3 (3): 229–241. doi:10.1677/erc.0.0030229. ISSN1351-0088.
↑ Ayub M, Levell MJ (August 1989). "The effect of ketoconazole related imidazole drugs and antiandrogens on [3H] R 1881 binding to the prostatic androgen receptor and [3H]5 alpha-dihydrotestosterone and [3H]cortisol binding to plasma proteins". Journal of Steroid Biochemistry. 33 (2): 251–255. doi:10.1016/0022-4731(89)90301-4. PMID2788775.
↑ Kemppainen JA, Wilson EM (July 1996). "Agonist and antagonist activities of hydroxyflutamide and Casodex relate to androgen receptor stabilization". Urology. 48 (1): 157–163. doi:10.1016/S0090-4295(96)00117-3. PMID8693644.
↑ Furr BJ (1997). "Relative potencies of flutamide and 'Casodex'". Endocrine-Related Cancer. 4 (2): 197–202. doi:10.1677/erc.0.0402197 (inactive 12 July 2025). 'Casodex' has been used in both models; in the Shionogi mammary tumour, an independent study by Darbre & King (1990) shows that a 1000-fold excess of 'Casodex' completely inhibits the response to 5α-DHT. Inspection of the data shown by Luo et al. suggests that at least a 10 000-fold excess of 'Casodex' and around a 3500-fold excess of hydroxyflutamide is required to achieve complete inhibition of 5α-DHT-stimulated growth.{{cite journal}}: CS1 maint: DOI inactive as of July 2025 (link)
↑ Gentile V, Panebianco V, Sciarra A (11 April 2014). Multidisciplinary Management of Prostate Cancer: The Role of the Prostate Cancer Unit. Springer Science & Business Media. pp.106–. ISBN978-3-319-04385-2. The standard castrate level is <50 ng/dl. It was defined more than 40 years ago, when testosterone level testing was limited. However, current testing methods using chemiluminescence have found that the mean value of testosterone after surgical castration is 15 ng/dL.
1 2 3 Boccardo F, Rubagotti A, Conti G, Potenzoni D, Manganelli A, Del Monaco D (October 2005). "Exploratory study of drug plasma levels during bicalutamide 150 mg therapy co-administered with tamoxifen or anastrozole for prophylaxis of gynecomastia and breast pain in men with prostate cancer". Cancer Chemotherapy and Pharmacology. 56 (4): 415–420. doi:10.1007/s00280-005-1016-1. PMID15838655. S2CID23014567.
1 2 Chabner BA, Longo DL (8 November 2010). Cancer Chemotherapy and Biotherapy: Principles and Practice. Lippincott Williams & Wilkins. pp.679–680. ISBN978-1-60547-431-1. From a structural standpoint, antiandrogens are classified as steroidal, including cyproterone [acetate] (Androcur) and megestrol [acetate], or nonsteroidal, including flutamide (Eulexin, others), bicalutamide (Casodex), and nilutamide (Nilandron). The steroidal antiandrogens are rarely used.
↑ Wirth MP, Hakenberg OW, Froehner M (February 2007). "Antiandrogens in the treatment of prostate cancer". European Urology. 51 (2): 306–13, discussion 314. doi:10.1016/j.eururo.2006.08.043. PMID17007995.
1 2 3 4 5 6 Tyrrell CJ, Iversen P, Tammela T, Anderson J, Björk T, Kaisary AV, etal. (September 2006). "Tolerability, efficacy and pharmacokinetics of bicalutamide 300 mg, 450 mg or 600 mg as monotherapy for patients with locally advanced or metastatic prostate cancer, compared with castration". BJU International. 98 (3): 563–572. doi:10.1111/j.1464-410X.2006.06275.x. PMID16771791. S2CID41672303.
1 2 Schellhammer PF, Davis JW (March 2004). "An evaluation of bicalutamide in the treatment of prostate cancer". Clinical Prostate Cancer. 2 (4): 213–219. doi:10.3816/cgc.2004.n.002. PMID15072604.
↑ Labrie F (January 2015). "Combined blockade of testicular and locally made androgens in prostate cancer: a highly significant medical progress based upon intracrinology". The Journal of Steroid Biochemistry and Molecular Biology. 145: 144–156. doi:10.1016/j.jsbmb.2014.05.012. PMID24925260. S2CID23102323.
1 2 Crawford ED, Schellhammer PF, McLeod DG, Moul JW, Higano CS, Shore N, etal. (November 2018). "Androgen Receptor Targeted Treatments of Prostate Cancer: 35 Years of Progress with Antiandrogens". The Journal of Urology. 200 (5): 956–966. doi:10.1016/j.juro.2018.04.083. PMID29730201. S2CID19162538.
↑ Styne DM (6 December 2019). "Physiology and Disorders of Puberty". In Melmed S, Koenig RJ, Rosen CJ, Auchus RJ, Goldfine AB, Williams RH (eds.). Williams Textbook of Endocrinology (14ed.). Philadelphia, PA: Elsevier. pp.1023–1164. ISBN978-0-323-55596-8.
1 2 3 Bouchard P, Caraty A (15 November 1993). GnRH, GnRH Analogs, Gonadotropins and Gonadal Peptides. CRC Press. pp.455–456. ISBN978-0-203-09205-7. [...] when male levels of androgens are achieved in plasma, their effects on gonadotropin secretion are similar in women and men. [...] administration of flutamide in a group of normally-cycling women produced a clinical improvement of acne and hirsutism without any significant hormonal change. [...] All these data emphasize that physiological levels of androgens have no action on the regulation of gonadotropins in normal women. [...] Androgens do not directly play a role in gonadotropin regulation [in women].
↑ Moretti CG, Guccione L, Di Giacinto P, Cannuccia A, Meleca C, Lanzolla G, etal. (2016). "Efficacy and Safety of Myo-Inositol Supplementation in the Treatment of Obese Hirsute PCOS Women: Comparative Evaluation with OCP+Bicalutamide Therapy". Endocrine Reviews. 37 (2).[verification needed]
1 2 Fradet Y (February 2004). "Bicalutamide (Casodex) in the treatment of prostate cancer". Expert Review of Anticancer Therapy. 4 (1): 37–48. doi:10.1586/14737140.4.1.37. PMID14748655. S2CID34153031. In contrast, the incidence of diarrhea was comparable between the bicalutamide and placebo groups (6.3 vs. 6.4%, respectively) in the EPC program [71].
1 2 3 4 5 6 7 8 9 10 Tyrrell CJ, Denis L, Newling D, Soloway M, Channer K, Cockshott ID (1998). "Casodex 10-200 mg daily, used as monotherapy for the treatment of patients with advanced prostate cancer. An overview of the efficacy, tolerability and pharmacokinetics from three phase II dose-ranging studies. Casodex Study Group". European Urology. 33 (1): 39–53. doi:10.1159/000019526. PMID9471040. S2CID71758492. The increase in testosterone in the previously published studies, and those reported from these Phase II studies, suggests that the elevation of this androgen is insufficient to overcome the blocking effect on Casodex on the androgen receptors, as shown by sustained reduction in PSA with Casodex. All doses from 30 mg and above showed intrinsic activity in terms of reductions in PSA, with doses of 50 mg and above giving reductions of 90% or more. At doses in the range 100-200 mg, reductions were in the region of 95% for all three doses. This feature of Casodex is especially important in considering its potential as monotherapy for prostate cancer, although in combination with an LHRH analogue this would not be an issue since testosterone levels would be suppressed. [...] What was clear, however, were the significant objective and subjective responses seen with associated reduction in serum PSA. This occurred despite elevated levels of testosterone, which suggests that the increases in testosterone caused by negative feedback on the pituitary-hypothalamic-gonadal axis do not interfere significantly with the blocking action of Casodex at the prostate receptor sites.[excessivequote]
1 2 Eri LM, Haug E, Tveter KJ (March 1995). "Effects on the endocrine system of long-term treatment with the non-steroidal anti-androgen Casodex in patients with benign prostatic hyperplasia". British Journal of Urology. 75 (3): 335–340. doi:10.1111/j.1464-410X.1995.tb07345.x. PMID7537602.
1 2 3 4 Kennealey GT, Furr BJ (February 1991). "Use of the nonsteroidal anti-androgen Casodex in advanced prostatic carcinoma". The Urologic Clinics of North America. 18 (1): 99–110. doi:10.1016/S0094-0143(21)01397-5. PMID1992575.
↑ Verhelst J, Denis L, Van Vliet P, Van Poppel H, Braeckman J, Van Cangh P, etal. (October 1994). "Endocrine profiles during administration of the new non-steroidal anti-androgen Casodex in prostate cancer". Clinical Endocrinology. 41 (4): 525–530. doi:10.1111/j.1365-2265.1994.tb02585.x. PMID7525125. S2CID7880831.
1 2 Wadhwa VK, Weston R, Parr NJ (June 2011). "Bicalutamide monotherapy preserves bone mineral density, muscle strength and has significant health-related quality of life benefits for osteoporotic men with prostate cancer". BJU International. 107 (12): 1923–1929. doi:10.1111/j.1464-410X.2010.09726.x. PMID20950306. S2CID205543615.
↑ Lunglmayr G (1989). "Casodex (ICI 176,334), a new, non-steroidal anti-androgen. Early clinical results". Hormone Research. 32 (Suppl 1): 77–81. doi:10.1159/000181316. PMID2515147.
↑ Müller EE (6 December 2012). Peptides and Non Peptides of Oncologic and Neuroendocrine Relevance: From Basic to Clinical Research. Springer Science & Business Media. pp.231–. ISBN978-88-470-2085-6. Pure anti-androgens can be given as monotherapy in the attempt to avoid the side effects caused by androgen-suppressive therapies (loss of libido, impotency, osteoporosis, pathological fractures, decrease of muscle mass and tone, progressive anaemia, asthenia, and depression) (Tyrrell, 1992). The use of these compounds in patients with intact gonads induces a condition of hypergonadotrophic hypergonadism, which allows high circulating levels of testosterone to be maintained.
↑ Knuth UA, Hano R, Nieschlag E (November 1984). "Effect of flutamide or cyproterone acetate on pituitary and testicular hormones in normal men". The Journal of Clinical Endocrinology and Metabolism. 59 (5): 963–969. doi:10.1210/jcem-59-5-963. PMID6237116. Since FLU is devoid of intrinsic hormonal activity, its antiandrogenic property leads to increased serum testosterone (T) levels and elevated gonadotropin values. The effect of this unique endocrine situation, which may be described as "hypergonadotropic hypergonadism."
↑ Müderris II, Bayram F, Ozçelik B, Güven M (February 2002). "New alternative treatment in hirsutism: bicalutamide 25 mg/day". Gynecological Endocrinology. 16 (1): 63–66. doi:10.1080/gye.16.1.63.66. PMID11915584. S2CID6942048.
↑ Bahceci M, Tuzcu A, Canoruc N, Tuzun Y, Kidir V, Aslan C (2004). "Serum C-reactive protein (CRP) levels and insulin resistance in non-obese women with polycystic ovarian syndrome, and effect of bicalutamide on hirsutism, CRP levels and insulin resistance". Hormone Research. 62 (6): 283–287. doi:10.1159/000081973. PMID15542929. S2CID46261843.
↑ Melmed S (1 January 2016). Williams Textbook of Endocrinology. Elsevier Health Sciences. pp.752–. ISBN978-0-323-29738-7. GnRH analogues, both agonists and antagonists, severely suppress endogenous gonadotropin and testosterone production [...] Administration of GnRH agonists (e.g., leuprolide, goserelin) produces an initial stimulation of gonadotropin and testosterone secretion (known as a "flare"), which is followed in 1 to 2 weeks by GnRH receptor downregulation and marked suppression of gonadotropins and testosterone to castration levels. [...] To prevent the potential complications associated with the testosterone flare, AR antagonists (e.g., bicalutamide) are usually coadministered with a GnRH agonist for men with metastatic prostate cancer.399
↑ Asscheman H, Gooren LJ, Peereboom-Wynia JD (September 1989). "Reduction in undesired sexual hair growth with anandron in male-to-female transsexuals--experiences with a novel androgen receptor blocker". Clinical and Experimental Dermatology. 14 (5): 361–363. doi:10.1111/j.1365-2230.1989.tb02585.x. PMID2612040. S2CID45303518.
↑ Rao BR, de Voogt HJ, Geldof AA, Gooren LJ, Bouman FG (October 1988). "Merits and considerations in the use of anti-androgen". Journal of Steroid Biochemistry. 31 (4B): 731–737. doi:10.1016/0022-4731(88)90024-6. PMID3143862.
1 2 Jameson JL, de Kretser DM, Marshall JC, De Groot LJ (7 May 2013). Endocrinology Adult and Pediatric: Reproductive Endocrinology. Elsevier Health Sciences. ISBN978-0-323-22152-8. Archived from the original on 25 July 2014. Nonsteroidal antiandrogens (e.g., flutamide and nilutamide) are also used, but they increase gonadotropin secretion, causing increased secretion of testosterone and estradiol.119 The latter is desirable in this context, as it has feminizing effects.
↑ Bach PV, Najari BB, Kashanian JA (2016). "Adjunct Management of Male Hypogonadism". Current Sexual Health Reports. 8 (4): 231–239. doi:10.1007/s11930-016-0089-7. S2CID79220716.
↑ Santen RJ, Leonard JM, Sherins RJ, Gandy HM, Paulsen CA (December 1971). "Short- and long-term effects of clomiphene citrate on the pituitary-testicular axis". The Journal of Clinical Endocrinology and Metabolism. 33 (6): 970–979. doi:10.1210/jcem-33-6-970. PMID5135636. Increase in serum LH levels ranged from 200–700% during the initial 21 days of clomiphene administration but then plateaued. Serum FSH levels exhibited a similar plateau after 35 days, with maximum titers 70–360% over control. The range in serum testosterone increments after 7 and 51 days of clomiphene administration was similar to that observed in serum gonadotrophin levels.
↑ Martini L (2 December 2012). Clinical Neuroendocrinology. Elsevier. p.239. ISBN978-0-323-14429-2. From the studies of Santen et al. (1971), it seems that a longer period of administration (51 days in their study) would cause an even greater rise in FSH and LH (70–360% and 200–700%, respectively).
1 2 Blackledge GR (1996). "Clinical progress with a new antiandrogen, Casodex (bicalutamide)". European Urology. 29 (2 Suppl 2): 96–104. doi:10.1159/000473847. PMID8717470.
↑ Veldhuis JD, Urban RJ, Dufau ML (June 1992). "Evidence that androgen negative feedback regulates hypothalamic gonadotropin-releasing hormone impulse strength and the burst-like secretion of biologically active luteinizing hormone in men". The Journal of Clinical Endocrinology and Metabolism. 74 (6): 1227–1235. doi:10.1210/jcem.74.6.1592863. PMID1592863.
1 2 Metzger DL, Kerrigan JR (May 1993). "Androgen receptor blockade with flutamide enhances growth hormone secretion in late pubertal males: evidence for independent actions of estrogen and androgen". The Journal of Clinical Endocrinology and Metabolism. 76 (5): 1147–1152. doi:10.1210/jcem.76.5.8496305. PMID8496305.
1 2 3 Wibowo E, Schellhammer P, Wassersug RJ (January 2011). "Role of estrogen in normal male function: clinical implications for patients with prostate cancer on androgen deprivation therapy". The Journal of Urology. 185 (1): 17–23. doi:10.1016/j.juro.2010.08.094. PMID21074215.
1 2 Motofei IG, Rowland DL, Popa F, Kreienkamp D, Paunica S (July 2011). "Preliminary study with bicalutamide in heterosexual and homosexual patients with prostate cancer: a possible implication of androgens in male homosexual arousal". BJU International. 108 (1): 110–115. doi:10.1111/j.1464-410X.2010.09764.x. PMID20955264. S2CID45482984.
1 2 Wibowo E, Wassersug RJ (September 2013). "The effect of estrogen on the sexual interest of castrated males: Implications to prostate cancer patients on androgen-deprivation therapy". Critical Reviews in Oncology/Hematology. 87 (3): 224–238. doi:10.1016/j.critrevonc.2013.01.006. PMID23484454.
↑ Simpson ER, Jones ME (2007). "Of mice and men: the many guises of estrogens". Tissue-Specific Estrogen Action. Ernst Schering Foundation Symposium Proceedings. Vol.2006/1. pp.45–67. doi:10.1007/2789_2006_016. ISBN978-3-540-49547-5. PMID17824171.
↑ Gaudet M, Vigneault É, Foster W, Meyer F, Martin AG (January 2016). "Randomized non-inferiority trial of Bicalutamide and Dutasteride versus LHRH agonists for prostate volume reduction prior to I-125 permanent implant brachytherapy for prostate cancer". Radiotherapy and Oncology. 118 (1): 141–147. doi:10.1016/j.radonc.2015.11.022. PMID26702991. Dutasteride and Bicalutamide is a regimen of non-inferior efficacy to LHRH agonist based regimens for prostate volume reduction prior to permanent implant prostate brachytherapy. D + B has less sexual toxicity compared to LHRH agonists prior to implant and for the first 6 months after implant. D + B is therefore an option to be considered for prostate volume reduction prior to PIPB.
↑ Mahler C, Verhelst J, Denis L (May 1998). "Clinical pharmacokinetics of the antiandrogens and their efficacy in prostate cancer". Clinical Pharmacokinetics. 34 (5): 405–417. doi:10.2165/00003088-199834050-00005. PMID9592622. S2CID25200595. If used in monotherapy, libido and potency are largely preserved. Although the mechanisms to explain this are not completely understood, it seems that at the central level pure antiandrogens are unable to completely inhibit the effect of the increased amount of androgens.
1 2 Ricci F, Buzzatti G, Rubagotti A, Boccardo F (November 2014). "Safety of antiandrogen therapy for treating prostate cancer". Expert Opin Drug Saf. 13 (11): 1483–99. doi:10.1517/14740338.2014.966686. PMID25270521. S2CID207488100. Bone-sparing effects of antiandrogen monotherapy might be due to selective AR modulators, tissue-specific and androgen-responsive, not affected by antiandrogen therapy, resulting in testosterone still being active in bone during non-steroidal antiandrogen administration [90].
1 2 3 4 5 6 7 8 Lefort M, Díaz Curiel M, Carrascal MT, Méndez-Dávila C, de la Piedra C (2005). "Comparative effects of bicalutamide (Casodex) versus orchidectomy on bone mineral density, bone remodelling, and bone biomechanics in healthy rats". Urol Int. 74 (4): 301–7. doi:10.1159/000084427. PMID15897693. S2CID11782787.
1 2 3 4 Yamada Y, Takahashi S, Fujimura T, Nishimatsu H, Ishikawa A, Kume H, etal. (March 2008). "The effect of combined androgen blockade on bone turnover and bone mineral density in men with prostate cancer". Osteoporos Int. 19 (3): 321–7. doi:10.1007/s00198-007-0472-3. PMID17906826. S2CID20614043. In our study, BMD% Z score of the castration group was lower than that of the 'CAB with BL' group but not statistically significant. However, bone turnover markers in the castration group were significantly higher than levels in the 'CAB with BL' group, indicating that bone turnover is occurring at a greater rate in the castration group (Fig. 1, 2 and 3). [...] The theory of the 'selective androgen receptor modulator' (SARM) may additionally explain what is behind the suppression mechanism of bone turnover in the 'CAB with BL' group. Draper et al. reported that BL effects on osteoblast androgen receptors is similar to the way selective estrogen receptor modulators act on bone [18]. Lefort M. et al. also suggested that BL may effect bone the same way that selective estrogen receptor modulator (SERM)does [19]. He introduced an earlier report by Hobauer et al. [20] who showed that, hydroxyflutamide, another non-steroidal anti-androgens, inhibits the interleukin-6 production in an androgen-responsive human osteoblastic cell line, indicating that this antiandrogen may function as a SARM for the effects on the interleukin-6 production in osteoblasts. Since BL is also a family of non-steroidal anti-androgens, this compound could also have similar effects on bone. If this mechanism is true, BL administration could prevent bone loss even in situations when administration of LH-RH agonists or surgical castration is given concurrently.[excessivequote]
1 2 Chisamore MJ, Gentile MA, Dillon GM, Baran M, Gambone C, Riley S, etal. (October 2016). "A novel selective androgen receptor modulator (SARM) MK-4541 exerts anti-androgenic activity in the prostate cancer xenograft R-3327G and anabolic activity on skeletal muscle mass & function in castrated mice". J Steroid Biochem Mol Biol. 163: 88–97. doi:10.1016/j.jsbmb.2016.04.007. PMID27106747. S2CID12202170. Schmidt and colleagues recently reported that bicalutamide resembles a selective androgen receptor modulator (SARM) in that it partially regulates endogenous AR-responsive genes as effectively as DHT in AR(+) MDA-MB-453 breast cancer cells (18).
↑ Dalton JT, Taylor RP, Mohler ML, Steiner MS (December 2013). "Selective androgen receptor modulators for the prevention and treatment of muscle wasting associated with cancer". Curr Opin Support Palliat Care. 7 (4): 345–51. doi:10.1097/SPC.0000000000000015. PMID24189892. S2CID35120033. Enobosarm was discovered in 2004 as a hyper-myoanabolic SARM that dissociated the anabolic from androgenic effects of AR in terms of potency (ED50) and efficacy (Emax) [29]. Levator ani muscle weight was increased to 131 and 136% of intact controls in intact and castrated (maintenance mode) rats, respectively, without significant increases in ventral prostate and seminal vesicles weights. Importantly, increases in levator ani muscle weight were associated with increases in muscle strength (soleus) in rats. Enobosarm also exerted in-vivo osteoanabolic effects alone and synergistically with alendronate in terms of bone density, strength, and structure [30], which was explained by in-vitro mechanistic studies that demonstrated antiresorptive (osteoclast inhibition) and anabolic (osteoblast differentiation) effects [31].
↑ Mohler ML, Bohl CE, Narayanan R, He Y, Hwang DJ, Dalton JT, etal. (2008-08-20). "Nonsteroidal Tissue-Selective Androgen Receptor Modulators". Methods and Principles in Medicinal Chemistry. Wiley. pp.249–304. doi:10.1002/9783527623297.ch8. ISBN978-3-527-31872-8.
↑ Kolvenbag GJ (2001). "The Prostate Cancer Continuum: The Role of Bicalutamide (CasodexR)". The Prostate Journal. 3 (1): 2–13. doi:10.1046/j.1525-1411.2001.003001002.x.
1 2 3 4 5 6 7 Schmidt A, Meissner RS, Gentile MA, Chisamore MJ, Opas EE, Scafonas A, etal. (September 2014). "Identification of an anabolic selective androgen receptor modulator that actively induces death of androgen-independent prostate cancer cells". J Steroid Biochem Mol Biol. 143: 29–39. doi:10.1016/j.jsbmb.2014.02.005. PMID24565564. S2CID825635.
↑ Zhang X, Li X, Allan GF, Sbriscia T, Linton O, Lundeen SG, etal. (August 2007). "Design, synthesis, and in vivo SAR of a novel series of pyrazolines as potent selective androgen receptor modulators". Journal of Medicinal Chemistry. 50 (16): 3857–3869. doi:10.1021/jm0613976. PMID17636947. Compounds (S)-7c and (R)-7c were further tested in intact mature male rats to assess antagonist activity on endogenous testosterone. [...] For comparison, the androgen antagonists flutamide and bicalutamide reduced both prostate and levator ani weights by 51 to 67% at 30 mg/kg.
1 2 3 4 Allan G, Lai MT, Sbriscia T, Linton O, Haynes-Johnson D, Bhattacharjee S, etal. (January 2007). "A selective androgen receptor modulator that reduces prostate tumor size and prevents orchidectomy-induced bone loss in rats". J Steroid Biochem Mol Biol. 103 (1): 76–83. doi:10.1016/j.jsbmb.2006.07.006. PMID17049844. S2CID25283876. Administration of DHT to castrate rats prevented loss of body mass and induced a body composition profile comparable to that of intact rats. Both JNJ-26146900 and bicalutamide also prevented orchidectomy-induced loss of body weight and lean body mass, albeit less effectively than DHT. Both compounds stabilized lean mass and total body weight, but compound-treated rats did not gain weight. [...] JNJ-26146900 partially prevented the loss of lean body mass that occurred in orchidectomized rats over a period of 8 weeks (Fig. 5). [...] JNJ-26146900-treated rats lost a mean of 3 g, meaning that they prevented 88% of the lean body loss. Bicalutamide was also effective. Neither compound was as effective as DHT, which promoted a gain in lean mass of 2 g in the castrated animals.
1 2 3 Sieber PR, Keiller DL, Kahnoski RJ, Gallo J, McFadden S (June 2004). "Bicalutamide 150 mg maintains bone mineral density during monotherapy for localized or locally advanced prostate cancer". The Journal of Urology. 171 (6 Pt 1): 2272–6, quiz 2435. doi:10.1097/01.ju.0000127738.94221.da. PMID15126801.
1 2 Klotz L (April 2008). "Maximal androgen blockade for advanced prostate cancer". Best Pract Res Clin Endocrinol Metab. 22 (2): 331–40. doi:10.1016/j.beem.2008.01.004. PMID18471790. One study has shown that loss of bone mineral density with MAB is reduced compared to castration alone.32
1 2 Kim SH, Seong do H, Yoon SM, Choi YD, Song Y, Song H, etal. (April 2016). "Bone health and its correlates in Korean prostate cancer patients receiving androgen deprivation therapy". Eur J Oncol Nurs. 21: 197–204. doi:10.1016/j.ejon.2015.10.004. PMID26522218. Interestingly, we found that compared with GnRH agonist monotherapy, combined androgen blockade was beneficial for BMD at the spine and femur neck. While previous studies have shown that androgen blockade monotherapy for patients with prostate cancer maintains BMD (Sieber et al., 2004; Smith et al., 2005), the effect of combined androgen blockades on BMD is not yet well understood. Yamada et al. (2008) suggest the possibility of attenuation of bone loss when bicalutamide is used for combined androgen blockades in patients with prostate cancer (Yamada et al., 2008). In their study, BMD in the hormone-naïve group was significantly higher than in the GnRH agonist-alone group (p = 0.016), but not significantly different than in the combined androgen blockades group (p = 0.33). In our study, bicalutamide was also used for androgen blockade. This non-steroidal antiandrogen inhibits interleukin-6 production in an androgenresponsive osteoblastic cell line, indicating that it may function as a selective androgen receptor modulator (Hofbauer et al., 1999). Draper (2003) reported that the bicalutamide effects on osteoblast androgen receptors are similar to the effects of selective oestrogen receptor modulators on bone. If this mechanism is confirmed, combined androgen blockades may prevent bone loss in patients with prostate cancer during ADT.
↑ Ikeda K, Horie-Inoue K, Inoue S (July 2019). "Functions of estrogen and estrogen receptor signaling on skeletal muscle". The Journal of Steroid Biochemistry and Molecular Biology. 191 105375. doi:10.1016/j.jsbmb.2019.105375. PMID31067490. S2CID145024087.
↑ Kim J, Wu D, Hwang DJ, Miller DD, Dalton JT (October 2005). "The para substituent of S-3-(phenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethyl-phenyl)-propionamides is a major structural determinant of in vivo disposition and activity of selective androgen receptor modulators". J Pharmacol Exp Ther. 315 (1): 230–9. doi:10.1124/jpet.105.088344. PMID15987833. S2CID30799845.
↑ Jeffrey R (2012). Effects of the anti-androgen bicalutamide on prostate cancer cells (Thesis). ProQuest1266232486. A comparison of the effects of bicalutamide treatment and AR knockdown by siRNA in LNCaP cells, using a limited gene array, demonstrates that the two treatments induce differential effects on transcription 35, suggesting that bicalutamide does more than simply abrogate androgen action and may modulate the expression of a unique subset of genes through the AR. Alternatively, bicalutamide may have cellular targets in addition to the AR. Collectively, these data suggest that bicalutamide, when bound to the AR, does not prevent AR mediated transcription but may alter its activity or that bicalutamide may have cellular targets in addition to the AR. Evidence of the continued activity of AR in the presence of bicalutamide is supported by analysis of an array of AR binding compounds or Selective Androgen Receptor Modulators (SARMs), which indicate that binding to the AR produces unique sets of cellular responses through distinct alterations of AR structure 36. Although binding of the AR to DNA and/or cofactors may be altered in the presence of anti-androgens 32, 37, current data suggest that the anti-androgen bound AR retains some activity in transcriptional complexes.[pageneeded][excessivequote]
↑ Hodgson MC, Astapova I, Hollenberg AN, Balk SP (September 2007). "Activity of androgen receptor antagonist bicalutamide in prostate cancer cells is independent of NCoR and SMRT corepressors". Cancer Res. 67 (17): 8388–95. doi:10.1158/0008-5472.CAN-07-0617. PMID17804755. This result is not inconsistent with the current study because flutamide has substantial agonist activity for the mutant AR (T877A) expressed in LNCaP cells. Therefore, the agonist activities of both DHT and flutamide in these cells are dependent on the balance between coactivators and corepresssors, and on this basis, these ligands may be classified as selective AR modulators. In contrast, we conclude that bicalutamide lacks any substantial partial agonist activity, and its activity is therefore not dependent on corepressors or on coactivator-to-corepressor ratios. Importantly, these ratios may become significant if the bicalutamide-liganded AR in advanced prostate cancer cells can acquire the ability to more effectively recruit coactivators (through increased coactivator expression, AR or coactivator phosphorylation, or other modifications).
1 2 Reid P, Kantoff P, Oh W (1999). "Antiandrogens in prostate cancer". Invest New Drugs. 17 (3): 271–84. doi:10.1023/a:1006344807086. PMID10665479. S2CID12658622. The development of tissue selective antiandrogens, termed selective androgen receptor antagonists (SARM), and further clinical experience with antiandrogens in drug combinations that achieve androgen blockade without castrate levels of testosterone are active areas of research that may affect the use of antiandrogens in the future. The relative concentrations of the natural ligand to inhibitor at the target receptor is an important determinant of the effectiveness of competitive inhibition. It is believed that the increased testosterone levels caused by NSAA limits the ability to achieve total AR inhibition. Inhibition of the AR in peripheral tissues but not in the CNS could provide AR blockade without increased serum testosterone levels because the negative feedback of testosterone on gonadotropin release would not be interrupted. Hence the effort to develop SARM that block peripheral but not CNS androgen receptors. Initially, there was some hope that bicalutamide would functionally prove to be a SARM based on animal experiments that suggested preferential inhibition of peripheral versus central nervous system AR [92]. Unfortunately bicalutamide did not prove to be peripherally selective in humans.
↑ Zanardi S, Puntoni M, Maffezzini M, Bandelloni R, Branchi D, Argusti A, etal. (1 December 2006). "A biomarker trial of intermittent, low-dose bicalutamide in subjects at high risk for prostate cancer: Updated results". Cancer Epidemiol Biomarkers Prev. 15 (12 Supplement): A145. Testosterone (T), LH, E2 and SHBG levels increased on Bic, although only T changes on both doses and LH changes on Bic 100 mg were significantly different to controls (p<0.001). Changes in circulating hormones and biomarkers were evident after 3 months, and persisted through the 6th month of treatment. No change in circulating hormones or biomarkers was observed in the control group. [...] Treatment was well tolerated, although breast pain was recorded in 0/19 (0%), 8/25 (32%) and 14/22 (64%), and gynecomastia in 0/19 (0%), 11/25 (44%) and 11/22 (50%) of subjects on no treatment, Bic 50 or 100 mg, respectively.
1 2 Fourcade RO, McLeod D (March 2004). "Tolerability of Antiandrogens in the Treatment of Prostate Cancer". UroOncology. 4 (1): 5–13. doi:10.1080/1561095042000191655. Gynaecomastia and breast or nipple tenderness are expected pharmacological effects of non-steroidal antiandrogen monotherapy and occur both because of the increase in circulating oestrogens and the unopposed oestrogen action at the breast due to androgen receptor blockade [6].
↑ Anderson J (March 2003). "The role of antiandrogen monotherapy in the treatment of prostate cancer". BJU International. 91 (5): 455–461. doi:10.1046/j.1464-410X.2003.04026.x. PMID12603397. S2CID8639102. Gynaecomastia is an expected pharmacological effect of nonsteroidal antiandrogen monotherapy and is caused by an imbalance in the normal ratio of androgenic and oestrogenic activity [32]. This is a result of an increase in oestrogen levels (after peripheral aromatization of elevated serum testosterone levels) and direct antagonism of androgen action in breast tissue.
1 2 Di Lorenzo G, Autorino R, Perdonà S, De Placido S (December 2005). "Management of gynaecomastia in patients with prostate cancer: a systematic review". The Lancet. Oncology. 6 (12): 972–979. doi:10.1016/S1470-2045(05)70464-2. PMID16321765.
↑ Wassersug RJ, Oliffe JL (April 2009). "The social context for psychological distress from iatrogenic gynecomastia with suggestions for its management". The Journal of Sexual Medicine. 6 (4): 989–1000. doi:10.1111/j.1743-6109.2008.01053.x. PMID19175864. By themselves, the LH-RH agonists do not produce much gynecomastia (ie, estimates as low as 4.4%) [13], but in conjunction with the typically prescribed antiandrogens (flutamide, bicalutamide, and nilutamide), gynecomastia is more common (49–68%) [13].
↑ Howard BA, Gusterson BA (April 2000). "Human breast development". Journal of Mammary Gland Biology and Neoplasia. 5 (2): 119–137. doi:10.1023/A:1026487120779. PMID11149569. S2CID10819224. In the stroma, there is an increase in the amount of fibrous and fatty tissue, with the adult nonlactating breast consisting of 80% or more of stroma.
↑ Sperling MA (10 April 2014). Pediatric Endocrinology. Elsevier Health Sciences. pp.598–. ISBN978-1-4557-5973-6. Estrogen stimulates the nipples to grow, mammary terminal duct branching to progress to the stage at which ductules are formed, and fatty stromal growth to increase until it constitutes about 85% of the mass of the breast. [...] Lobulation appears around menarche, when multiple blind saccular buds form by branching of the terminal ducts. These effects are due to the presence of progesterone. [...] Full alveolar development normally only occurs during pregnancy under the influence of additional progesterone and prolactin.
↑ Hagisawa S, Shimura N, Arisaka O (June 2012). "Effect of excess estrogen on breast and external genitalia development in growth hormone deficiency". Journal of Pediatric and Adolescent Gynecology. 25 (3): e61 –e63. doi:10.1016/j.jpag.2011.11.005. PMID22206682. Estrogen stimulates growth of the nipples, progression of mammary duct branching to the stage at which ductiles are formed, and fatty stromal growth until it constitutes about 85% of the mass of the breast.
↑ Fody EP, Walker EM (1985). "Effects of drugs on the male and female reproductive systems". Annals of Clinical and Laboratory Science. 15 (6): 451–458. PMID4062226.
1 2 3 Morgante E, Gradini R, Realacci M, Sale P, D'Eramo G, Perrone GA, etal. (March 2001). "Effects of long-term treatment with the anti-androgen bicalutamide on human testis: an ultrastructural and morphometric study". Histopathology. 38 (3): 195–201. doi:10.1046/j.1365-2559.2001.01077.x. hdl:11573/387981. PMID11260298. S2CID36892099.
1 2 Bjerklund Johansen TE, Majak M, Nesland JM (March 1994). "Testicular histology after treatment with the new antiandrogen Casodex for carcinoma of the prostate. A preliminary report". Scandinavian Journal of Urology and Nephrology. 28 (1): 67–70. doi:10.3109/00365599409180473. PMID8009196.
↑ Jones HB, Betton GR, Bowdler AL, McFarquhar RL, Middleton BJ, Lunglmayr G (1994). "Pathological and morphometric assessment of testicular parameters in patients with metastatic prostate cancer following treatment with either the antiandrogen Casodex (ZM176,334) or bilateral orchidectomy". Urological Research. 22 (3): 191–195. doi:10.1007/BF00571849. PMID7992465. S2CID19540140.
↑ Lunglmayr G, Girsch E, Kuber W, Biegelmayr C (1988). Effect of a new non-steroidal antiandrogen (ICI 176,334) on hormone levels and testis morphology of patients with advanced prostate cancer (abstract 583). Proceedings of the 21st Congress of the International Society of Urology, October 9–14, 1988, Buenos Aires, Argentina.
↑ Abdulrahman AS, Mustafa IA (2019). "Impact of bicalutamide, an anti-androgen on rat testis". Zanco Journal of Pure and Applied Sciences. 31 (2). doi:10.21271/ZJPAS.31.2.12 (inactive 12 July 2025).{{cite journal}}: CS1 maint: DOI inactive as of July 2025 (link)
↑ Chandolia RK, Weinbauer GF, Behre HM, Nieschlag E (March 1991). "Evaluation of a peripherally selective antiandrogen (Casodex) as a tool for studying the relationship between testosterone and spermatogenesis in the rat". The Journal of Steroid Biochemistry and Molecular Biology. 38 (3): 367–375. doi:10.1016/0960-0760(91)90109-I. PMID1848993. S2CID19423860.
↑ Chandolia RK, Weinbauer GF, Simoni M, Behre HM, Nieschlag E (November 1991). "Comparative effects of chronic administration of the non-steroidal antiandrogens flutamide and Casodex on the reproductive system of the adult male rat". Acta Endocrinologica. 125 (5): 547–555. doi:10.1530/acta.0.1250547. PMID1759544.
1 2 Jones CA, Reiter L, Greenblatt E (2016). "Fertility preservation in transgender patients". International Journal of Transgenderism. 17 (2): 76–82. doi:10.1080/15532739.2016.1153992. S2CID58849546. Traditionally, patients have been advised to cryopreserve sperm prior to starting cross-sex hormone therapy as there is a potential for a decline in sperm motility with high-dose estrogen therapy over time (Lubbert et al., 1992). However, this decline in fertility due to estrogen therapy is controversial due to limited studies.
1 2 3 4 5 6 Payne AH, Hardy MP (28 October 2007). The Leydig Cell in Health and Disease. Springer Science & Business Media. pp.422–431. ISBN978-1-59745-453-7. Estrogens are highly efficient inhibitors of the hypothalamic-hypophyseal-testicular axis (212–214). Aside from their negative feedback action at the level of the hypothalamus and pituitary, direct inhibitory effects on the testis are likely (215,216). [...] The histology of the testes [with estrogen treatment] showed disorganization of the seminiferous tubules, vacuolization and absence of lumen, and compartmentalization of spermatogenesis.
1 2 Salam MA (2003). Principles & Practice of Urology: A Comprehensive Text. Universal-Publishers. pp.684–. ISBN978-1-58112-412-5. Estrogens act primarily through negative feedback at the hypothalamic-pituitary level to reduce LH secretion and testicular androgen synthesis. [...] Interestingly, if the treatment with estrogens is discontinued after 3 yr. of uninterrupted exposure, serum testosterone may remain at castration levels for up to another 3 yr. This prolonged suppression is thought to result from a direct effect of estrogens on the Leydig cells.
1 2 3 4 5 Foster WR, Car BD, Shi H, Levesque PC, Obermeier MT, Gan J, etal. (April 2011). "Drug safety is a barrier to the discovery and development of new androgen receptor antagonists". The Prostate. 71 (5): 480–488. doi:10.1002/pros.21263. PMID20878947. S2CID24620044.
↑ Saqib U, Savai R, Liu D, Banerjee S, Baig MS (February 2019). "Drug repositioning as an effective therapy for protease-activated receptor 2 inhibition". Journal of Cellular Biochemistry. 120 (2): 1522–1526. doi:10.1002/jcb.27334. PMID30370939. S2CID53099338.
↑ Zia MK, Siddiqui T, Ali SS, Ahsan H, Khan FH (December 2018). "Exploring the interaction of anti-androgen drug-bicalutamide with human alpha-2-macroglobulin: A biophysical investigation". International Journal of Biological Macromolecules. 120 (Pt B): 2285–2292. doi:10.1016/j.ijbiomac.2018.08.117. PMID30149080. S2CID52094869.
1 2 3 4 Weber GF (22 July 2015). Molecular Therapies of Cancer. Springer. pp.318–. ISBN978-3-319-13278-5. Compared to flutamide and nilutamide, bicalutamide has a 2-fold increased affinity for the Androgen Receptor, a longer half-life, and substantially reduced toxicities. Based on a more favorable safety profile relative to flutamide, bicalutamide is indicated for use in combination therapy with a Gonadotropin Releasing Hormone analog for the treatment of advanced metastatic prostate carcinoma.
↑ Cockshott ID, Plummer GF, Cooper KJ, Warwick MJ (October 1991). "The pharmacokinetics of Casodex in laboratory animals". Xenobiotica; the Fate of Foreign Compounds in Biological Systems. 21 (10): 1347–1355. doi:10.3109/00498259109043209. PMID1796611.
1 2 Sharma K, Pawar GV, Giri S, Rajagopal S, Mullangi R (December 2012). "Development and validation of a highly sensitive LC-MS/MS-ESI method for the determination of bicalutamide in mouse plasma: application to a pharmacokinetic study". Biomedical Chromatography. 26 (12): 1589–1595. doi:10.1002/bmc.2736. PMID22495777.
1 2 3 Cantarini M, Fuhr R, Morris T (2006). "Pharmacokinetics of two novel bicalutamide formulations in healthy male volunteers". Pharmacology. 77 (4): 171–178. doi:10.1159/000094599. PMID16847400. S2CID35010482.
↑ Allan G, Sbriscia T, Linton O, Lai MT, Haynes-Johnson D, Bhattacharjee S, etal. (June 2008). "A selective androgen receptor modulator with minimal prostate hypertrophic activity restores lean body mass in aged orchidectomized male rats". J Steroid Biochem Mol Biol. 110 (3–5): 207–13. doi:10.1016/j.jsbmb.2007.10.012. PMID18502117. S2CID25985722. For comparison, the androgen antagonists flutamide and bicalutamide increased intact levels of both FSH (twofold) and T (fivefold).
↑ Soloway MS, Schellhammer PF, Smith JA, Chodak GW, Vogelzang NJ, Kennealey GT (December 1995). "Bicalutamide in the treatment of advanced prostatic carcinoma: a phase II noncomparative multicenter trial evaluating safety, efficacy and long-term endocrine effects of monotherapy". The Journal of Urology. 154 (6): 2110–2114. doi:10.1016/S0022-5347(01)66709-0. PMID7500470.
↑ Mason M (August 2006). "What implications do the tolerability profiles of antiandrogens and other commonly used prostate cancer treatments have on patient care?". Journal of Cancer Research and Clinical Oncology. 132 (Suppl 1): S27 –S35. doi:10.1007/s00432-006-0134-4. PMID16896883. S2CID19685819.
1 2 Colabufo NA, Pagliarulo V, Berardi F, Contino M, Inglese C, Niso M, etal. (December 2008). "Bicalutamide failure in prostate cancer treatment: involvement of Multi Drug Resistance proteins". European Journal of Pharmacology. 601 (1–3): 38–42. doi:10.1016/j.ejphar.2008.10.038. PMID18992739.
↑ Schinkel AH (April 1999). "P-Glycoprotein, a gatekeeper in the blood-brain barrier". Advanced Drug Delivery Reviews. 36 (2–3): 179–194. doi:10.1016/s0169-409x(98)00085-4. PMID10837715.
↑ Fromm MF (February 2000). "P-glycoprotein: a defense mechanism limiting oral bioavailability and CNS accumulation of drugs". International Journal of Clinical Pharmacology and Therapeutics. 38 (2): 69–74. doi:10.5414/cpp38069. PMID10706193.
↑ Chu X, Bleasby K, Evers R (March 2013). "Species differences in drug transporters and implications for translating preclinical findings to humans". Expert Opin Drug Metab Toxicol. 9 (3): 237–252. doi:10.1517/17425255.2013.741589. PMID23256482.
↑ Syvänen S, Lindhe O, Palner M, Kornum BR, Rahman O, Långström B, etal. (March 2009). "Species differences in blood-brain barrier transport of three positron emission tomography radioligands with emphasis on P-glycoprotein transport". Drug Metab Dispos. 37 (3): 635–643. doi:10.1124/dmd.108.024745. PMID19047468.
↑ Knoben JE, Anderson PO (22 August 2001). Handbook of clinical drug data. McGraw Hill Professional. ISBN978-0-07-138942-6. With an oral dose of 50 mg/day, bicalutamide attains a peak serum level of 8.9 mg/L (21 μmol/L) 31 hr after a dose at steady state. CI of (R)-bicalutamide is 0.32 L/hr. The active (R)-enantiomer of bicalutamide is oxidized to an inactive metabolite, which, like the inactive (S)-enantiomer, is glucuronidated and cleared rapidly by elimination in the urine and feces.165
↑ Cockshott ID (2004). "Bicalutamide: clinical pharmacokinetics and metabolism". Clinical Pharmacokinetics. 43 (13): 855–878. doi:10.2165/00003088-200443130-00003. PMID15509184. S2CID29912565. These data indicate that direct glucuronidation is the main metabolic pathway for the rapidly cleared (S)-bicalutamide, whereas hydroxylation followed by glucuronidation is a major metabolic pathway for the slowly cleared (R)-bicalutamide.
1 2 McKillop D, Boyle GW, Cockshott ID, Jones DC, Phillips PJ, Yates RA (November 1993). "Metabolism and enantioselective pharmacokinetics of Casodex in man". Xenobiotica; the Fate of Foreign Compounds in Biological Systems. 23 (11): 1241–1253. doi:10.3109/00498259309059435. PMID8310708.
1 2 Cockshott ID, Cooper KJ, Sweetmore DS, Blacklock NJ, Denis L (1990). "The pharmacokinetics of Casodex in prostate cancer patients after single and during multiple dosing". European Urology. 18 (3): 10–17. doi:10.1159/000463972. PMID2094606.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.




 Testosterone levels with 10, 30, and 50 mg/day bicalutamide monotherapy in men with prostate cancer. [89]
Testosterone levels with 10, 30, and 50 mg/day bicalutamide monotherapy in men with prostate cancer. [89] Testosterone levels with 10 to 200 mg/day bicalutamide monotherapy in men with prostate cancer. [85]
Testosterone levels with 10 to 200 mg/day bicalutamide monotherapy in men with prostate cancer. [85] Estradiol levels with 10, 30, and 50 mg/day bicalutamide monotherapy in men with prostate cancer. [89]
Estradiol levels with 10, 30, and 50 mg/day bicalutamide monotherapy in men with prostate cancer. [89] Estradiol levels with 10 to 200 mg/day bicalutamide monotherapy in men with prostate cancer. [85]
Estradiol levels with 10 to 200 mg/day bicalutamide monotherapy in men with prostate cancer. [85]