Congenital insensitivity to pain (CIP), also known as congenital analgesia, is one or more extraordinarily rare conditions in which a person cannot feel physical pain. The conditions described here are separate from the HSAN group of disorders, which have more specific signs and cause. Because feeling physical pain is vital for survival, CIP is an extremely dangerous condition. It is common for people with the condition to die in childhood due to injuries or illnesses going unnoticed. Burn injuries are among the more common injuries.

Erythromelalgia or Mitchell's disease is a rare vascular peripheral pain disorder in which blood vessels, usually in the lower extremities or hands, are episodically blocked, then become hyperemic and inflamed. There is severe burning pain and skin redness. The attacks are periodic and are commonly triggered by heat, pressure, mild activity, exertion, insomnia or stress. Erythromelalgia may occur either as a primary or secondary disorder. Secondary erythromelalgia can result from small fiber peripheral neuropathy of any cause, polycythemia vera, essential thrombocythemia, hypercholesterolemia, mushroom or mercury poisoning, and some autoimmune disorders. Primary erythromelalgia is caused by mutation of the voltage-gated sodium channel α-subunit gene SCN9A.

Sodium channels are integral membrane proteins that form ion channels, conducting sodium ions (Na+) through a cell's membrane. They belong to the superfamily of cation channels.

Sodium channel protein type 4 subunit alpha is a protein that in humans is encoded by the SCN4A gene.

Sodium channel protein type 5 subunit alpha, also known as NaV1.5 is an integral membrane protein and tetrodotoxin-resistant voltage-gated sodium channel subunit. NaV1.5 is found primarily in cardiac muscle, where it mediates the fast influx of Na+-ions (INa) across the cell membrane, resulting in the fast depolarization phase of the cardiac action potential. As such, it plays a major role in impulse propagation through the heart. A vast number of cardiac diseases is associated with mutations in NaV1.5 (see paragraph genetics). SCN5A is the gene that encodes the cardiac sodium channel NaV1.5.
Small fiber peripheral neuropathy is a type of peripheral neuropathy that occurs from damage to the small unmyelinated and myelinated peripheral nerve fibers. These fibers, categorized as C fibers and small Aδ fibers, are present in skin, peripheral nerves, and organs. The role of these nerves is to innervate some skin sensations and help control autonomic function. It is estimated that 15–20 million people in the United States have some form of peripheral neuropathy.
T-type calcium channels are low voltage activated calcium channels that become inactivated during cell membrane hyperpolarization but then open to depolarization. The entry of calcium into various cells has many different physiological responses associated with it. Within cardiac muscle cell and smooth muscle cells voltage-gated calcium channel activation initiates contraction directly by allowing the cytosolic concentration to increase. Not only are T-type calcium channels known to be present within cardiac and smooth muscle, but they also are present in many neuronal cells within the central nervous system. Different experimental studies within the 1970s allowed for the distinction of T-type calcium channels from the already well-known L-type calcium channels. The new T-type channels were much different from the L-type calcium channels due to their ability to be activated by more negative membrane potentials, had small single channel conductance, and also were unresponsive to calcium antagonist drugs that were present. These distinct calcium channels are generally located within the brain, peripheral nervous system, heart, smooth muscle, bone, and endocrine system.

Sodium channel, voltage-gated, type XI, alpha subunit also known as SCN11A or Nav1.9 is a voltage-gated sodium ion channel protein which is encoded by the SCN11A gene on chromosome 3 in humans. Like Nav1.7 and Nav1.8, Nav1.9 plays a role in pain perception. This channel is largely expressed in small-diameter nociceptors of the dorsal root ganglion and trigeminal ganglion neurons, but is also found in intrinsic myenteric neurons.
Paroxysmal extreme pain disorder originally named familial rectal pain syndrome, is a rare disorder whose most notable features are pain in the mandibular, ocular and rectal areas as well as flushing. PEPD often first manifests at the beginning of life, perhaps even in utero, with symptoms persisting throughout life. PEPD symptoms are reminiscent of primary erythromelalgia, as both result in flushing and episodic pain, though pain is typically present in the extremities for primary erythromelalgia. Both of these disorders have recently been shown to be allelic, both caused by mutations in the voltage-gated sodium channel NaV1.7 encoded by the gene SCN9A. A different mutation in the SCN9A ion channel causes congenital insensitivity to pain.

Sodium channel protein type 1 subunit alpha (SCN1A), is a protein which in humans is encoded by the SCN1A gene.

Sodium channel protein type 2 subunit alpha, is a protein that in humans is encoded by the SCN2A gene. Functional sodium channels contain an ion conductive alpha subunit and one or more regulatory beta subunits. Sodium channels which contain sodium channel protein type 2 subunit alpha are sometimes called Nav1.2 channels.

Sodium channel protein type 8 subunit alpha also known as Nav1.6 is a membrane protein encoded by the SCN8A gene. Nav1.6 is one sodium channel isoform and is the primary voltage-gated sodium channel at each node of Ranvier. The channels are highly concentrated in sensory and motor axons in the peripheral nervous system and cluster at the nodes in the central nervous system.

Nax is a protein that in humans is encoded by the SCN7A gene. It is a sodium channel alpha subunit expressed in the heart, the uterus and in glial cells of mice. It has low similarity to all nine other sodium channel alpha subunits (Nav1.1–1.9).
Nav1.8 is a sodium ion channel subtype that in humans is encoded by the SCN10A gene.

Acid-sensing ion channels (ASICs) are neuronal voltage-insensitive sodium channels activated by extracellular protons permeable to Na+. ASIC1 also shows low Ca2+ permeability. ASIC proteins are a subfamily of the ENaC/Deg superfamily of ion channels. These genes have splice variants that encode for several isoforms that are marked by a suffix. In mammals, acid-sensing ion channels (ASIC) are encoded by five genes that produce ASIC protein subunits: ASIC1, ASIC2, ASIC3, ASIC4, and ASIC5. Three of these protein subunits assemble to form the ASIC, which can combine into both homotrimeric and heterotrimeric channels typically found in both the central nervous system and peripheral nervous system. However, the most common ASICs are ASIC1a and ASIC1a/2a and ASIC3. ASIC2b is non-functional on its own but modulates channel activity when participating in heteromultimers and ASIC4 has no known function. On a broad scale, ASICs are potential drug targets due to their involvement in pathological states such as retinal damage, seizures, and ischemic brain injury.
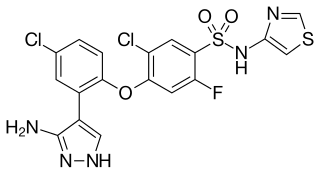
PF-05089771 is a selective, small-molecule Nav1.7 and Nav1.8 voltage-gated sodium channel blocker under development by Pfizer as a novel analgesic. As of June 2014, it has completed phase II clinical trials for wisdom tooth removal and primary erythromelalgia.
Voltage-gated sodium channels (VGSCs), also known as voltage-dependent sodium channels (VDSCs), are a group of voltage-gated ion channels found in the membrane of excitable cells (e.g., muscle, glial cells, neurons, etc.) with a permeability to the sodium ion Na+. They are the main channels involved in action potential of excitable cells.

Stephen George Waxman is an American neurologist and neuroscientist. He served as Chairman of the Department of Neurology at Yale School of Medicine, and Neurologist-in-Chief at Yale-New Haven Hospital from 1986 until 2009. As of 2023, he is the Bridget Flaherty Professor of Neurology, Neurobiology, and Pharmacology at Yale University. He founded the Yale University Neuroscience & Regeneration Research Center in 1988 and is its director. He previously held faculty positions at Harvard Medical School, MIT, and Stanford Medical School. He is also visiting professor at University College London. He is the editor-in-chief of The Neuroscientist.
Protoxin-II, also known as ProTx-II, PT-II or β/ω-TRTX-Tp2a, is a neurotoxin that inhibits certain voltage-gated calcium and voltage-gated sodium channels. This toxin is a 30-residue disulfide-rich peptide that has unusually high affinity and selectivity toward the human Nav1.7. channel.
Phlotoxin is a neurotoxin from the venom of the tarantula Phlogiellus that targets mostly voltage-sensitive sodium channels and mainly Nav1.7. The only non-sodium voltage-sensitive channel that is inhibited by Phlotoxin is Kv3.4. Nav1.4 and Nav1.6 seem to be Phlotoxin-1-sensitive to some extent as well.





